
基于最大期望算法的蛋白质交互关系识别
2018-08-21 01:59蔡松成
计算机技术与发展 2018年8期
蔡松成,牛 耘
(南京航空航天大学 计算机科学与技术学院,江苏 南京 211106)
0 引 言
随着人们对文本中分子途径和分子交互关系等信息需求的不断增加,蛋白质交互作用关系(protein-protein interaction,PPI)的自动抽取在分子生物学领域变得越来越重要。PPI是指细胞内两个蛋白质之间的交互作用,这种交互作用环环相扣,深刻影响着整个细胞生理作用的调节。起初生物医学领域的专家手工地从医学文献中收集这些信息录入统一格式的数据库中,如HPRD[1]、IntAc[2]、MINT[3]和BIND[4]等。然而随着生物医学文献的急剧增加,新的蛋白质之间的关系也在产生。手工录入蛋白质之间的交互信息显然远不能满足实际需要,因此自动地从医学文献中抽取PPI已经成为一项重要的研究内容。
在此背景下,基于自然语言处理的PPI自动识别技术正在快速发展并已取得了很大的进展。目前PPI识别是采用有监督的机器学习方法,以单句为依据来识别句子之间的交互关系,需要大量人工标注的数据,代价高昂,所以将远监督的思想运用到PPI识别上,解决了训练数据不足的问题。但是由于远监督思想的缺陷,引入了大量噪音,影响现阶段PPI识别的精度。针对这个问题,采用一种基于最大期望算法的多实例多标记学习(multi-instance multi-label,MIML)方法来进行蛋白质交互关系的识别,有效消除了签名档中噪音对交互关系识别的影响。
1 相关工作
目前,用于从生物医学文献中抽取PPI的技术主要包括:基于同现的方法[5]、基于规则的方法和基于机器学习[6-8]的方法。基于同现的方法通过统计两个蛋白质在句子中的共现频率来判断是否存在交互关系,识别结果召回率高但精确度低;基于规则的方法可以取得较高的精确度但是召回率较低,而且通过手动建立规则的方法需要大量的人力物力,且制定的规则只适用于某些特定领域的数据,无法普遍应用。
随着机器学习的流行,研究者们越来越多地采用基于机器学习的方法进行PPI的识别。基于机器学习的方法主要包括两大类:基于特征的方法和基于核函数的方法。基于特征的方法从标注有交互关系的句子中抽取重要特征,包括词汇特征、语法特征和语义特征,建立模型来判断蛋白质之间的交互关系[9-10]。基于核函数的方法首先深入研究句子结构,通过设计核函数进一步利用句子结构表示(如字符串序列、句法依赖或句法分析)上的隐含特征,然后使用支持核函数的分类器进行PPI关系的识别。Haussler D[11]提出了针对离散结构的卷积核;Lodhi H等[12]将特征空间特定长度词语子序列的内积作为函数的计算方式,提出了字符串核;Bunescu R C等[13]提出了最短依赖路径核,将句子以树的形式表示,用两个实体之间的最短路径表示实体之间的关系。然而目前利用机器学习方法来进行PPI关系识别一般都是以句子为单位,分析一句话中出现的任意一对蛋白质对之间是否存在交互关系。这种方式能够在句子级别上提供蛋白质对交互关系的描述和证据,但是也存在一定的局限性。这种方式所需的训练集要求对每一个句子中出现的每一对蛋白质是否存在交互关系进行标注,当训练语料不足时,PPI关系识别的效果会大打折扣。但人工标注大规模文本需要耗费大量的人力物力。
针对这些不足,文中试图采用远监督思想来进行PPI关系的抽取。远监督方法已经用于关系识别领域,远监督思想假设如果两个实体之间存在某种关系,那么包含这两个实体的所有句子都在一定程度上表达了这种关系。基于上述假设,远监督通过将知识库中的实体和训练语料文本中的实体进行匹配,产生大量带标注的训练数据,避免了人工标注数据的繁重劳动。对于PPI关系识别,同样存在标注数据不足的问题,所以可以将远监督[14-15]方法运用到PPI关系抽取上。
但是基于远监督方法的PPI识别也存在一个问题。对于有交互关系的蛋白质对事实上并非其签名档中的所有句子都表达了该蛋白质对的交互关系,其中很多句子是不表达交互关系的,从而这部分数据成为了训练过程中的噪音,最终会影响蛋白质对交互关系的识别结果。
针对远监督的PPI抽取方法存在的问题,文中采用一种基于最大期望算法的多实例多标记的学习方法。多实例多标记是一种新型的关系抽取的学习框架[16],在该框架中,每个对象由多个实例描述,同时对象可以拥有多个类别标记,这个框架尤其适用于多义性的对象。多实例多标记学习框架已被成功应用于图像文本分类[17]、视频标注[18]、基因图像识别[19]等任务中,既充分利用了蛋白质对签名档的信息,同时又改善了利用远监督思想来标记签名档中的句子带来的噪音问题。在此基础上又对特征加以改进,有效消除了其他蛋白质对目标蛋白质对交互关系识别的影响。
2 基于最大期望算法的PPI识别
基于最大期望算法的多实例多标记学习方法,是在基于远监督方法的基础上,从大规模生物医学文献中搜索得到的蛋白质对签名档中提取特征,构建向量空间模型(vector space model,VSM)。在此基础上引入隐变量,将蛋白质对的签名档和标签构建为多实例多标记的学习框架,利用最大期望算法迭代地消除噪音。最终采用监督学习的方法来预测未知蛋白质对的交互关系。
2.1 关系提取
PubMed数据库作为建立PPI网络重要的数据来源,收录了超过一千八百万篇生物医学文献摘要。从PubMed数据中获取蛋白质对签名档的过程包括:
(1)调用PubMed数据库提供的接口,搜索包含目标蛋白质对的摘要。
(2)使用伊利诺州大学Urbana-Champaign分校认知计算研究组开发的句子识别工具来识别摘要集合中的句子,保留包含目标蛋白质对的句子作为签名档的内容。
最终每一个目标蛋白质对都会有一个包含多个句子的集合与之对应,这个句子集合即为蛋白质对的签名档,接下来将签名档作为蛋白质对交互关系的特征来源进行处理。
2.2 特征表示
实验中使用到了两个逻辑回归分类器来进行PPI关系的识别。一个是对蛋白质对签名档中的句子进行交互关系判断的句子级分类器,另一个是对蛋白质对进行分类的顶层分类器。两个分类器的主要差别在于特征的表示上,句子级分类器利用提取得到的句子的语言学特征进行分类,而顶层分类器通过当前签名档中句子的分类结果形成特征进行分类。句子级分类器特征的形成主要是选取训练集中所有句子中重要的单词特征作为向量的每一维。具体处理过程为:首先对句子进行分词,去除无意义的标点符号以及停用词;然后选取句子中出现在两个目标蛋白质之间的单词,以及第一个目标蛋白质左边2个单词和第二个目标蛋白质右边2个单词;最终将这些单词作为句子中蛋白质对的上下文特征来构建向量空间模型。若在句子的上下文特征中出现了某个特征词,则在向量中对应于出现特征词的某一维用1记录,否则用0记录。
对于顶层分类器中蛋白质对的实际交互关系,采取签名档中判断为有交互关系的句子数占签名档中所有句子的比例作为特征构建一维向量。
2.3 多实例多标记学习模型
在该模型中,对于训练集中的每一个蛋白质对,都有已知的唯一标记,即有无交互关系,但对于签名档中的每一个句子并不知道其真实的标记。所以,引入一个隐变量z来代表句子的标记。z=non-interactive表示在该句中目标蛋白质对之间没有交互关系;z=interactive表示目标蛋白质对之间存在交互关系。对于PPI关系抽取中的关系是互补的,两个蛋白质之间的关系就分为有交互和无交互两种。在该模型中,如图1所示,由两层构成,包含一个对蛋白质对签名档中的句子进行分类的句子级二元分类器(z分类器)和一个对蛋白质对进行分类的顶层二元分类器(y分类器)。
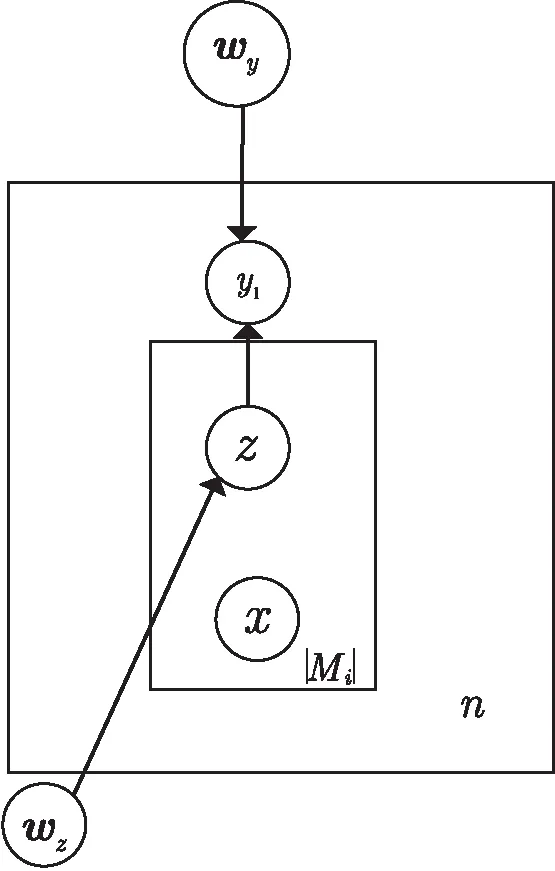
图1 多实例多标记学习框架
图中,n表示蛋白质对的数目;Mi表示第i对蛋白质对签名档的数目;x表示输入的一个句子;wz表示z分类器的权重向量;wy表示y分类器的权重向量。
2.3.1 训 练
由于蛋白质对签名档中的句子标记是未知的,而最大期望算法是估计隐变量的有效方法,所以文中采用最大期望算法来训练多实例多标记的学习框架。最大期望算法主要由M步和E步构成,M步训练句子级分类器(z分类器)和顶层分类器(y分类器),E步根据得到的两个分类器来更新句子的标记,经过多次迭代使句子的标记更加接近于真实的标记。
在以下的公式中,向量zi代表第i个蛋白质对所有句子的标记构成的一个向量;yi用来表示第i个蛋白质对的标记,用公式表示为:
(1)
其中,Pi为关系正例,表示第i对蛋白质对具有的关系;Ni是关系负例,表示第i对蛋白质对不具有的关系。
文中使用最大期望算法来最大化极大似然函数的下界,也就是说最大化数据库中每个蛋白质对的联合概率,得到:
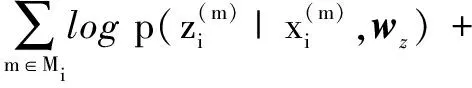
(2)
E-step:在此步骤,对于每个蛋白质对的签名档,给定蛋白质对的标记集合,以及目前模型学习得到的z分类器和y分类器的权重向量,推断出蛋白质对句子级别的分类结果。
(3)
通过近似化,将向量z进行拆分,分开考虑每个句子的分类结果。对于每个蛋白质对i=1,2,…,n中的每个句子m∈Mi,计算:
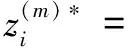
(4)
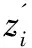
M-step:此步骤利用E-step得到的句子分类结果zi通过最大化似然函数的下界,得到对应的wz和wy。实际上就是通过学习来更新句子级和蛋白质对级权重参数,具体公式如下:
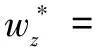
(5)
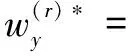
(6)
2.3.2 预 测
(1)对于一个给定的蛋白质对,首先预测其签名档中句子的分类结果。
(7)
(2)利用顶层分类器来决定该蛋白质对是否具有交互关系。
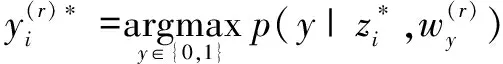
(8)
2.3.3 实 现
初始化:由于最大期望算法并不是全局最优算法,因此初始值的设置对最后的结果有着重要的影响。在该模型中,初始值为签名档中句子的类别分布zi。利用原始的签名档数据来训练一个分类器,然后通过此分类器对签名档中的句子进行分类,将分类结果作为初始值zi。
2.4 特征改进
通过对目标蛋白质对签名档数据的观察,发现在包含目标蛋白质对的同一个句子的描述中往往还存在其他蛋白质,这些蛋白质可能会对目标蛋白质交互关系的判断造成影响。基于这个原因,需要对句子级分类器原始的特征加以改进。
文中利用一个生物医学文本命名实体识别工具ABNER来识别句子中其他蛋白质的名称。ABNER在NLPBA和BioCreative语料库上进行训练,在两个语料库上识别的F值分别达到了72.6%和69.9%。
通过观察蛋白质对的签名档,可以发现描述交互作用的句子中经常会出现bind、interact、activate、inhibit、down-regulate等表示蛋白质交互作用的单词。这些单词通常被认为是识别蛋白质交互关系的关键词。关键词对于蛋白质交互关系识别尤为重要,已经作为线索运用到基于模式匹配的PPI抽取方法中。文中选择关键词作为一维特征对原有特征加以改进,采用的关键词集合利用了Joshua M.Temkin[20]提出的关键词列表。
观察以下描述蛋白质交互关系的句子:
#arnt# mRNA appeared to be slightly but significantly down-regulated by
用##标注出来的是两个目标蛋白质,而用
在保留2.1节所有特征的基础上,又新增了5个特征来对句子级分类器进行改进。首先对签名档中的每一个句子,抽取出第一个蛋白质左边的7个单词和第二个蛋白质右边的7个单词以及两个蛋白质中间的所有单词。然后将第一个蛋白质左边和第二个蛋白质右边是否有关键字和其他蛋白质的名称以及目标蛋白质中间有没有其他蛋白质作为5维特征添加到原有特征中,权重采用二值权重,若有则置为1,否则置为0。增加了这5维特征后,以第一个蛋白质左边的两个特征为例,若出现了关键词和其他蛋白质,则很有可能表示的是其他蛋白质和第一个目标蛋白质之间的交互关系。
3 实 验
3.1 实验数据及设置
采用的训练数据来自于现有的PPI数据库,无需额外的人工标注。将有交互关系的蛋白质对视为正样例,无交互的视为负样例。实验中有交互关系的蛋白质对是直接从HPRD数据库中查询获取,并且只保留被PubMed数据库中一篇以上摘要包含的那些蛋白质对。而对于无交互关系的蛋白质对,采用生物医学领域常用方法,将蛋白质随机组合成蛋白质对,去除已被HPRD数据库包含的蛋白质对以及未被PubMed数据库记载的蛋白质对。以两个待考察的蛋白质为查询条件,通过PubMed数据库的应用程序接口查询目标蛋白质对的文献摘要,然后对摘要文本集合进行处理,找出包含目标蛋白质对的句子,形成签名档。最终总共得到有交互关系和无交互关系的蛋白质对分别为576对和578对,合计1 154对。
实验采用的结果性能评价指标是当前PPI抽取系统主要使用的三个指标:精确度(precision=TP/(TP+FP))、召回率(recall=TP/(TP+FN))和F值(F-Score=2P×R/(P+R))。为了避免简单应用模型而产生过拟合问题,利用五折交叉验证来评估模型的性能。将原始数据按照蛋白质对平均划分为5折,将每个子集数据分别做一次验证集,其余的4组子集数据作为训练集,这样会得到5个模型,用这5个模型最终验证集的平均性能作为评价整个方法性能的指标。
3.2 实验结果及分析
为了比较使用原始特征和改进后特征的实验结果,以第一折数据为例,取最大期望算法迭代的前六次(迭代6次以后实验结果基本趋向局部最优解),结果如表1、表2所示。
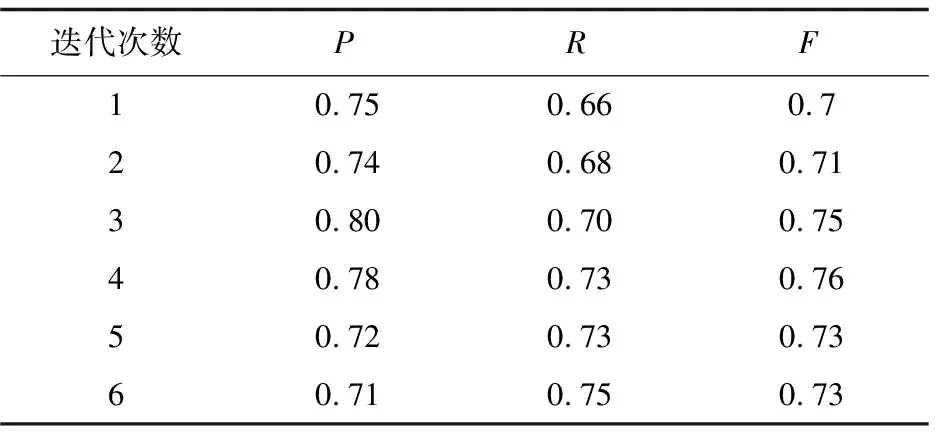
表1 采用原始特征的识别结果
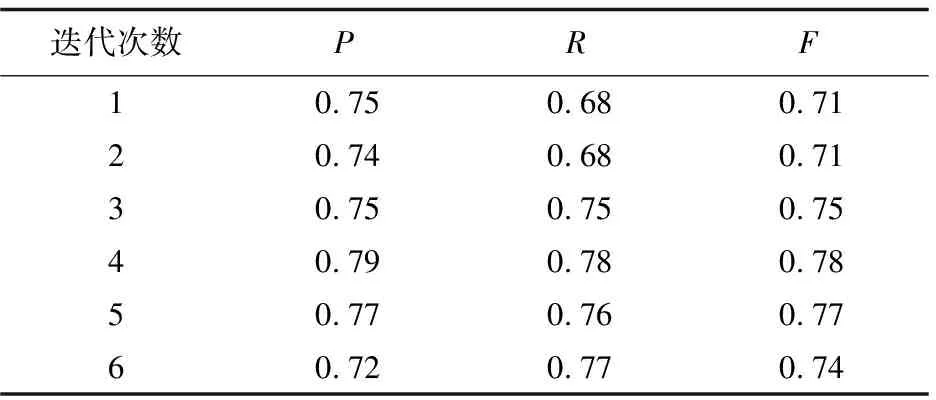
表2 采用改进特征的识别结果
从这两张表可以发现,随着迭代次数的增加,采用改进以后的特征在精确度、召回率和F值上都有明显提高。最终结果是要把五折数据识别的平均结果作为该模型PPI识别的性能,如表3所示。
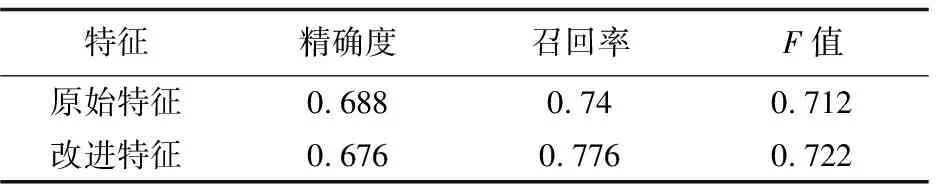
表3 五折交叉验证识别结果比较
从上述识别结果发现,对特征加以改进后,识别的准确率虽然稍有下降,但是召回率提高了3.6%,整体F值提高1%。说明改进后,算法考虑了其他蛋白质对目标蛋白质识别的影响,使模型取得了更好的性能。
4 结束语
由于基于远监督的PPI抽取方法存在大量噪音问题,文中采用基于最大期望算法的多实例多标记学习框架,同时在此基础上对特征加以改进,消除了签名档中其他蛋白质对目标蛋白质对交互关系判断的影响。实验结果表明,该方法取得了更高的识别精度。
下一步将利用蛋白质对签名档中包含的丰富信息对句子级分类器得到的结果进行改进,使句子级的分类更加准确,从而能进一步提高PPI识别的效果。
猜你喜欢
肝博士(2022年3期)2022-06-30
电子产品世界(2022年4期)2022-04-21
海外星云(2021年9期)2021-10-14
计算机系统应用(2021年2期)2021-02-23
电子技术与软件工程(2019年18期)2019-11-18
意林·全彩Color(2019年9期)2019-11-13
新城乡(2018年8期)2018-09-03
新城乡(2017年12期)2018-01-04
新城乡(2017年9期)2017-09-26
电子技术与软件工程(2017年14期)2017-09-08
