
消肿止痛液有效部位软膏的制备工艺及其质量标准研究
2018-11-05 08:02李晓
中国民族民间医药 2018年19期
李晓
1.银川市中医医院,宁夏 银川 750001;2.宁夏医科大学,宁夏 银川 750001
痔疮是肛肠科多发病,宁夏回族自治区地处西北,人们喜食牛羊肉、善饮酒等生活习惯导致痔疮发病率较高。据流行病学研究[1],该地区痔疮患病率高达40.06%。消肿止痛液(批准文号:宁药制字Z20140005)是银川市中医医院肛肠科临床使用二十多年的医院制剂,主要由地榆、黄柏、大黄等八味中药组成,用于治疗各种原因所致的痔疮初期及外痔、混合痔、肛裂和痔疮术后的消肿止痛,疗效确切[2]。我院制剂中心对其进行了洗液制备工艺[3]和薄层鉴别研究。随着生活节奏的加快,外用洗剂携带不方便,使用方法繁琐以及存储困难,不能满足患者需求,软膏制剂使用和携带方便,患者依从性好,因对消肿止痛液进行二次开发,旨在开发疗效确切,使用便捷的软膏制剂。前期通过对消肿止痛液的各部位成分进行富集,药效筛选得到有效部位,本论文将对有效部位软膏的制备工艺和质量标准进行研究。
1 仪器与材料
1.1 仪器 TU-1901型紫外分光光度计(北京普析通用仪器有限责任公司);XA105型电子分析天平(瑞士梅特勒-托利多仪器有限公司);YP4002型电子天平(上海越平科学仪器有限公司);JJ-1型精密增力电动搅拌器(金坛市城西峥嵘试验仪器厂);DZTW型调温电热套(北京市永光明医疗仪器厂);TGL-15B型高速离心机(上海安亭科学仪器厂)。
1.2 材料 消肿止痛液有效部位(银川市中医医院制剂中心实验室自制);硬脂酸(批号:101820150901,湖南尔康制药股份有限公司);羟苯乙酯(批号:103620170605,湖南尔康制药股份有限公司);三乙醇胺(批号:20170401,成都华邑药用辅料制造有限责任公司);甘油(批号:000120161004,湖南尔康制药股份有限公司);没食子酸(批号:110831-201605,纯度90.8%)、盐酸小檗碱(批号:110713-201212)、大黄素(批号:110756-201512)、大黄对照药材(批号:121249-201304)、黄柏对照药材(批号:121510-201606)均购于中国食品药品检定研究院。硅胶G薄层板(青岛海洋化工厂分厂)。所用试剂均为分析纯。
2 方法与结果
2.1 基质类型考察及药物准备 消肿止痛液有效部位软膏用于肛周局部给药,根据给药部位特点,肛周皮肤湿性重,并易产生分泌物[4],故选用O/W乳膏基质,不仅可吸附分泌物,还具有易于涂布和清洗的特点。参考《宁夏医院制剂规范》收载的“乳膏基质1号”进行基质优化。消肿止痛液有效部位药物细粉,过六号筛,备用。
2.2 乳化工艺考察[5]
2.2.1 试验方法与质量评价指标
2.2.1.1 离心试验高度比(Y1) 按正交试验设计制备软膏,分别称取样品20 g,置于内径为1 cm离心管中,以4000 r/min的转速离心30 min,取出,测量离心管中分离出的油层高度(Ho)、水层高度(Ha)及总高度(Ht),计算高度比(Y1)。计算公式:Y1=(Ho+Ha)/Ht
2.2.1.2 高温试验高度比(Y2) 按正交试验设计制备软膏,分别称取样品20g,置于内径为1 cm的试管中,于60℃水浴静置5 h,测量离心管中分离出的油层高度(Ho)、水层高度(Ha)及总高度(Ht),计算高度比(Y2)。计算公式同上。
2.2.1.3 综合评分 将Y1、Y2归一化为评价指标Y进行综合评分。两个指标的权重系数均为0.5,当软膏中油、水两相完全分层时,Y1或Y2值为1,Y1和Y2越小越好,即综合评分(Y)=[(1-Y1i)×0.5+(1-Y2i)×0.5]×100。
2.2.1.4 正交设计 根据前期预试验结果,采用L9(34)正交设计,以药物与基质比例(A)、乳化时间(B)和乳化温度(C)为影响因素,以离心试验高度比(Y1)和高温试验高度比(Y2)综合评分,并结合软膏的外观形状对其质量进行评价,优选消肿止痛有效部位软膏乳化工艺。
2.2.2 试验结果与数据分析 按“2.2.1”项下方法进行离心试验和高温试验,正交试验因素水平表见表1,正交试验结果见表2,方差分析见表3。
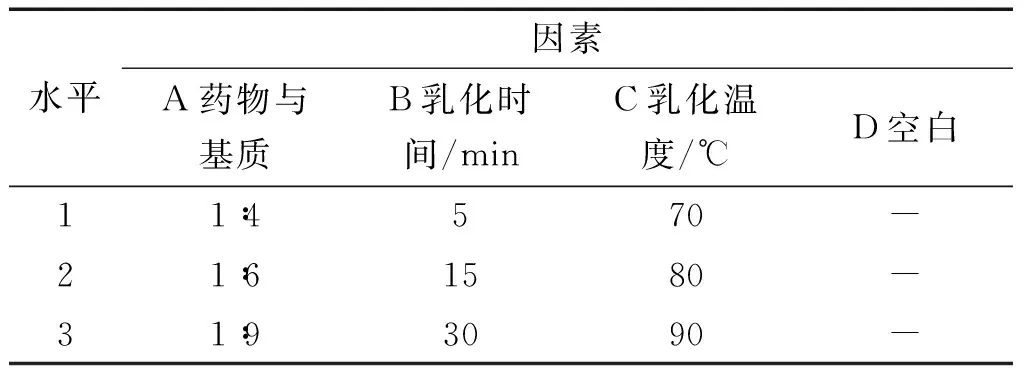
表1 正交试验因素水平表

表2 正交试验结果表

表3 方差分析表
注:F0.05(2,2)=19.0;F0.01(2,2)=99.0。
综合评分越高,表明软膏的稳定性越好,表3方差分析结果显示A因素对离心试验和高温试验结果有统计学意义,且A3>A2>A1,即药物与基质比第3水平1∶9最为适宜;B和C两个因素试验结果无统计学意义,且B3>B2>B1,C3>C2>C1,因此软膏乳化时间第3水平即30 min,加热温度为90 ℃,考虑实际生产温度控制,故温度控制在80 ℃较为合适。由此可知消肿止痛液有效部位软膏的最佳制备工艺为硬脂酸、石蜡、液体石蜡80 ℃共融作为油相;三乙醇胺、甘油、羟苯乙酯溶液(5%)、水共同加热至80 ℃作为水相,同时将药物细粉与基质按1∶9加入少量乙醇超声溶解,趁热加入水相,水相倒入油相,搅拌30 min,放冷至室温即得。
2.3 验证试验 按上述制备工艺平行制备3批消肿止痛液有效部位软膏,处方量为100 g,并按“2.2.1”项下方法进行离心试验和高温试验,从两相的分层情况、外观(颜色均匀度、软硬度及细腻程度)评价软膏制备工艺的合理性。结果表明,3份样品经高温、离心试验后均无分层,外观各项均符合要求。
2.4 大黄的鉴定[6]取软膏5.0 g,加入40%乙醇40 mL,加热回流1 h,滤过,滤液蒸干,残渣用1%氢氧化钠溶液调节pH至9~10,用乙酸乙酯振摇萃取2次,每次20 mL,弃去乙酸乙酯液,水层加入盐酸2 mL,用乙酸乙酯振摇提取2次,每次20 mL,合并乙酸乙酯液,蒸干,残渣用甲醇1 mL溶解,作为供试品溶液。按处方称取缺大黄的其他药材,分离得到有效部位药物,并按软膏制备工艺制成缺大黄的阴性样品,同法制备缺大黄的阴性对照溶液。另取大黄对照药材0.10 g,同法制成对照药材溶液。再称取大黄素,用甲醇配制成65.23 μg/mL对照品溶液。照薄层色谱法(中国药典2015年版四部通则0502)试验,吸取上述四种溶液5 μL,分别点于同一硅胶G薄层板上,以石油醚(60~90℃)-乙酸乙酯-甲酸(15∶5∶1)为展开剂,展开,取出,晾干,置紫外灯(365 nm)下检视,再置于氨蒸气中观察斑点颜色变化。结果,供试品色谱中在与对照药材色谱对应处,可见5个斑点显橙黄色荧光;在与对照品色谱对应处,可见相同颜色的斑点;用氨蒸气熏蒸后,可见斑点显红,阴性无干扰,见图1。

2.5 黄柏的鉴别[7-8]供试品溶液制备方法同“2.4”项下第一次乙酸乙酯振摇萃取后即合并提取液,蒸干,残渣用甲醇1 mL溶解,作为供试品溶液,同法制备缺黄柏的阴性对照溶液,另取黄柏对照药材0.10 g,同法制成对照药材溶液。再称取盐酸小檗碱,用甲醇配制成55.63 μg/mL对照品溶液。照薄层色谱法(中国药典2015年版四部通则0502)试验,吸取上述四种溶液5 μL,分别点于同一硅胶G薄层板上,以乙酸乙酯-丙酮-甲酸-水(10∶5∶1∶1)为展开剂,展开,取出,晾干,置紫外灯(365 nm)下检视。结果,供试品色谱中在与对照药材色谱对应处,可见黄绿色荧光斑点;在与对照品色谱对应处,可见相同颜色的斑点;阴性无干扰,见图2。

2.6 总鞣质含量测定[9-12]
2.6.1 溶液配制
2.6.1.1 磷钼钨酸试液的配制 取钨酸钠100 g,钼酸钠25 g,加水700 mL使溶解,加盐酸100 mL,磷酸50 mL,加热回流10 h,放冷,再加硫酸锂150 g,水50 mL和溴0.2 mL,煮沸除去残留的溴(约15 min)。冷却,加水稀释至1000 mL,滤过,即得。另外,磷钼钨酸试液不得显绿色(如放置后变为绿色,可加溴0.2 mL,煮沸出去多余的溴,可恢复原色。)
2.6.1.2 29%碳酸钠溶液的配制 称取无水碳酸钠29 g,溶于100 mL蒸馏水中,即得。(不溶解可水浴加热并不断搅拌至溶解,需现用现配。)
2.6.1.3 对照品溶液的配制 精密称取没食子酸对照品0.0504 g(纯度90.8%),置于100 mL棕色容量瓶中,加水溶解并稀释至刻度,精密量取5 mL,置50 mL棕色量瓶中,用水稀释至刻度,摇匀,即得45.76 μg/mL对照品溶液。
2.6.2 标准曲线的制备 精密量取对照品溶液1,2,4,6,8,10 mL,分别置50 mL棕色量瓶中,各加入磷钼钨酸试液2 mL,再分別加水23,22,20,18,16,14 mL,用29%碳酸钠溶液稀释至刻度,摇匀,放置30 min以相应的试剂为空白,照紫外-可见分光光度法(通则0401),在760 nm的波长处测定吸光度,以吸光度为纵坐标,浓度为横坐标,绘制标准曲线。线性回归方程为:Y=100.65x+0.3108(r=0.9996),没食子酸在0.0229~0.2290 mg吸光度与浓度呈良好的线性关系。
2.6.3 供试品溶液的制备[13]取供试品1.00 g,精密称定至100 mL棕色容量瓶,加水在80 ℃水浴中加热2 h并时时振摇,放冷,置冰浴中冷却1 h,迅速用滤膜(0.45 μm)滤过,精密量取5 mL,置100 mL棕色量瓶中,用水稀释至刻度,摇匀,即得。
2.6.4 总酚的测定 精密量取“2.6.3”项下供试品溶液4 mL,置50 mL棕色量瓶中,加入磷钼钨酸试液2 mL,加水20 mL,用29%碳酸钠溶液稀释至刻度,摇匀,放置30 min以不加供试品的试剂为空白依法测定吸光度,从“2.6.2”项下标准曲线中读出供试品溶液中没食子酸的浓度(μg/mL),计算,即得。
2.6.5 不被吸附的多酚的测定 精密量取供试品溶液25 mL,加至已盛有干酪素0.6 g的100 mL具塞锥形瓶中,密塞,置30 ℃水浴中保温1 h,时时振摇,取出,放冷,摇匀,滤过,弃去初滤液,精密量取续滤液4 mL,置50 mL棕色量瓶中,后同“2.6.4”项下操作相同,测得没食子酸的浓度(μg/mL),计算,即得。
2.6.6 总鞣质的测定 总鞣质的含量=总酚量-不被吸收多酚量
2.6.7 专属性考察 取供试品溶液按“2.6.4”和“2.6.5”项下方法制备样品,在190~800 nm波长范围内进行全波长扫描,结果在760 nm有最大吸收,与对照品相同,表明其他成分对鞣质测定无干扰。
2.6.8 精密度考察 精密量取对照品溶液4 mL,置50 mL棕色容量瓶中,加磷钼钨酸试液2 mL,加水20 mL,用29%碳酸钠溶液定容,放置30 min,以相应试剂为空白,平行测定吸光度值5次,计算RSD为0.06%,表明该方法精密度良好。
2.6.9 稳定性考察 取同一供试品溶液,分别于0,2,4,6,8,10,12 h测定总鞣质的含量,结果总鞣质的平均含量为24.85 mg/g,计算RSD为1.96%,表明所制备的供试品溶液在12 h内测定稳定性较好。
2.6.10 重复性考察 取同一批次样品6份,按“2.6.4”和“2.6.5”项下总酚和不被吸收多酚的方法制备,测定总鞣质的含量,结果总鞣质的平均含量为25.63 mg/g,计算RSD为1.99%,表明该方法重复性良好。
2.6.11 加样回收率试验 精密称取同批已知总鞣质含量的软膏样品6份,每份0.50 g,分别精密加入没食子酸对照品12.80 mg,按供试品溶液的方法制备,测定总鞣质的含量,结果见表4。结果表明,平均回收率为93.45%,RSD为2.69%,因此该方法样品处理方法可行。

表4 加样回收率试验结果
2.6.12 样品的含量测定 取“2.3”项下的制备3个不同批次软膏,精确称取1.00 g,每批次各3份,按“2.6.4”和“2.6.5”项下总酚和不被吸收多酚的方法制备,测定总鞣质的含量,结果见表5。3个批次软膏的含量为24.34~27.10 mg/g,RSD为0.64~2.23%。

表5 样品含量测定结果
3 讨论
研究制备软管为O/W型,硬脂酸与三乙醇胺作用生成的硬脂酸三乙醇胺一价皂,为阴离子型乳化剂,所制得的软膏pH约为8左右,刺激性较钾皂、钠皂小,成品较细腻有光泽但化学稳定性较差。根据文献资料[14],药物的量及成分性质影响药物的稳定性,主要有两个影响过程:第一,随时间而分层,大部分主药可能会迁移到与其极性相似的相体系中,造成主药在软膏中分布不均匀;第二,粒度的增加,制备过程中强剪切力形成的多相微粒体系随时间增加而丧失表面能,逐渐合并聚集成大颗粒。研究表明[15],乳状液的液滴大小明显随乳化温度而改变,所以乳化温度宜控制在70~90 ℃较为合适。在预试验过程中发现,乳化时间对软膏的乳化影响较为明显。因此选用药物与基质比例、乳化时间和乳化温度三个因素优化软膏制备工艺,解决该软膏稳定性差的问题,得到优化制备工艺参数。
软膏制备工艺时,药物为40%乙醇洗脱部位,在水中溶解性较差,因此加入少量乙醇(可对皮肤可产生刺激性,只能加少量)超声溶解后与水相搅拌混合均匀,使药物混悬在水相中。羟苯乙酯在软膏中的防腐剂。因用量小,直接加固体粉末不易均匀,用醇溶液使分散均匀。鞣质是一类复杂的具有沉淀蛋白质性质的水溶性多元酚类化合物,见光易分解,故本实验鞣质的含量测定过程中应避光操作且使用棕色瓶存储溶液。在总鞣质的测定中,软膏基质有一定影响,采用冰浴冷却,快速过滤膜的方法去除基质干扰。
综上,该软膏的制备是为了满足临床需要对原有医院制剂进行剂型开发,采用分离试验和高温试验评估软膏的成型工艺,确保软膏质量稳定同时建立软膏质量标准,为该制剂的二次开发打下基础。后期将按照《中国药典》2015版软膏项下检查指标开展微生物检查等相关检查试验,加快开发进度。
猜你喜欢
恋爱婚姻家庭·养生版(2020年7期)2020-08-10
食品研究与开发(2020年4期)2020-02-29
中成药(2017年7期)2017-11-22
中国现代医学杂志(2017年26期)2017-11-16
中国公路(2017年17期)2017-11-09
中国民族民间医药·上半月(2017年2期)2017-03-09
药学研究(2015年11期)2015-12-19
中国医疗美容(2015年1期)2015-07-12
食品工业科技(2014年13期)2014-03-11
筑路机械与施工机械化(2014年4期)2014-03-01
