
小蛋白质富集法鉴定酿酒酵母“漏检蛋白”
2018-12-07 01:04何崔同张瑶姜颖徐平
生物工程学报 2018年11期
何崔同,张瑶,姜颖,徐平
小蛋白质富集法鉴定酿酒酵母“漏检蛋白”
何崔同1,2,张瑶2,3,姜颖1,2,徐平1,2
1 安徽医科大学,安徽 合肥 230032 2 军事科学院军事医学研究院辐射医学研究所 蛋白质组学国家重点实验室 国家蛋白质科学中心 (北京) 北京蛋白质组研究中心,北京 102206 3 中山大学 生命科学学院,广东 广州 510275

何崔同, 张瑶, 姜颖, 等. 小蛋白质富集法鉴定酿酒酵母“漏检蛋白”. 生物工程学报, 2018, 34(11): 1860–1869.He CT, Zhang Y, Jiang Y, et al. Identification of missing proteins in Saccharomyces cerevisiae bysmall protein-based enrichment. Chin J Biotech, 2018, 34(11): 1860–1869.
小蛋白质 (Small proteins,SPs) 是由小开放阅读框 (Short open reading frames,sORFs) 编码长度小于100个氨基酸的多肽。研究发现小蛋白质参与了基因表达调控、细胞信号转导和代谢等重要生物学过程。然而,生命体中大多数的已注释小蛋白质尚缺少蛋白水平存在的实验证据,被称为漏检蛋白 (Missing proteins,MPs)。小蛋白质的高效鉴定是其功能研究的前提,也有助于挖掘“漏检蛋白”。文中采用小蛋白质富集策略鉴定到72个酵母小蛋白质,验证9个“漏检蛋白”,发现低分子量、高疏水性、膜结合、弱密码子使用偏性及不稳定性是蛋白漏检的主要原因,对进一步的技术优化具有指导意义。
蛋白质组学,小蛋白质,富集,漏检蛋白
小蛋白质 (Small proteins,SPs) 是由小开放阅读框 (Short open reading frames,sORFs) 编码长度小于100个氨基酸 (Amino acid,AA) 的多肽。小蛋白质在多数生命过程中发挥着重要的 作用,如蛋白互作与酶活性调节[1]、细胞信号 转导和细胞间通讯[2],以及基因表达的转录后调控[3]等。
然而,据UniProt数据库所示,目前几乎所有生命体中大多数已注释小蛋白质尚缺少蛋白水平存在的实验证据和相关功能研究,这类蛋白质被称为漏检蛋白 (Missing proteins,MPs)。UniProt数据库将所有已注释蛋白质存在证据 (Protein existence,PE) 分为5个水平,从PE1 (蛋白质水平实验证据) 到PE5 (无任何证据),其中漏检蛋白被定义为PE2 (转录组水平的实验证据)、PE3 (依据序列同源性推断的证据) 和PE4 (基于基因预测得到的证据)[4]。漏检蛋白研究在2012年已被人类染色体蛋白质组计划 (Chromosome-centric Human Proteome Project,C-HPP) 列为主要任务之一[5],但在其他生命体中尚未探及。
小蛋白质的有效鉴定是功能研究的前提,小蛋白质鉴定技术的发展不仅有助于从蛋白质水平验证漏检蛋白,而且为进一步漏检蛋白功能研究提供了技术手段。与基于荧光蛋白[6]和亲和富集标签[7]的生化方法相比,基于质谱鉴定的技术可以在蛋白质组水平高通量准确鉴定蛋白质且可以避免标签的引入对小蛋白质结构和功能的可能干扰[8]。小蛋白质的低丰度以及复杂体系中高丰度大蛋白质的信号掩盖是造成小蛋白质质谱鉴定困难的最直接原因[9]。
本研究以酿酒酵母为研究对象,采用小蛋白质富集策略来提高小蛋白质覆盖度。最终,我们共鉴定到72个小蛋白质,并成功验证9个漏检蛋白(Q02820、A5Z2X5、Q2V2P4、P38127、P40086、P40086、Q3E762、Q8TGU7、Q2V2P2)。小蛋白质和漏检蛋白特殊理化、生物学性质及其质谱鉴定规律的揭示对小蛋白质和漏检蛋白鉴定技术的进一步优化具有重要的指导意义。
1 材料与方法
1.1 材料
1.1.1 菌株
酿酒酵母JMP024[10]为本实验室保存。
1.1.2 主要试剂
葡萄糖 (Dextrose)、尿素 (Urea)、碳酸氢铵(NH4HCO3) 均为购自国药集团化学试剂有限公司的分析纯试剂;酵母粉 (Yeast extract)、蛋白胨 (Bacto-peptone) 均购自OXOID公司;叠氮钠(NaN3)、二硫苏糖醇 (Dithiothreitol,DTT)、碘乙酰胺 (Iodoacetamide,IAA) 均购自AMRESCO公司;2-巯基乙醇 (β-mercaptoethanol,β-ME)、聚乙二醇辛基苯基醚 (Triton X-100)、甲酸(Formic acid,FA)、色谱级乙腈 (Acetonitrile,ACN)均购自Sigma-Aldrich公司;质谱级胰蛋白酶(Trypsin) 为实验室自制试剂[11-12]。
1.1.3 主要仪器
LTQ Orbitrap Velos质谱仪为Thermo Fisher Scientific公司产品;超高压纳升级高效液相色谱仪(Ultra-high Performance Liquid Chromatography) 为Waters公司产品。
1.2 方法
1.2.1 细胞培养
将JMP024接种于YPD培养基 (1%酵母粉,2% 蛋白胨,2% 葡萄糖) 中,30 ℃振荡培养至对数中期 (600=1.5),离心并收集菌体,0.01% 叠氮钠洗涤菌体后于–80 ℃保存。
1.2.2 小蛋白质富集和蛋白质组样品制备
向上述收集的62(41.3 mL) 菌体中加入适量的裂解液Ⅰ (0.05 mol/L盐酸,0.1% 2-巯基乙醇,0.05%聚乙二醇辛基苯基醚)[9],玻璃珠涡旋振荡破碎 (工作30 s,冰浴30 s,10个循环)。细胞裂解后4 ℃、17 000×离心20 min,收集沉淀。沉淀用裂解液Ⅰ洗涤3次,离心取沉淀。将上述沉淀重悬于适量裂解液Ⅱ (8 mol/L尿素,0.05 mol/L 碳酸氢铵,0.005 mol/L碘乙酰胺)[13],4 ℃、17 000×离心20 min,收集上清。采用Xu灰度定量法[14]进行蛋白质定量。取80 μg蛋白于PCR管中,加入1 mol/L二硫苏糖醇至终浓度0.005 mol/L,45 ℃温浴30 min。加0.5 mol/L碘乙酰胺至终浓度0.01 mol/L,室温避光孵育30 min。还原、烷基化后的蛋白样品采用12% Tricine-SDS- PAGE短胶分离。待样品入分离胶3 cm时停止 电泳。再经考马斯亮蓝G250染色、脱色后,将 0–3.4 kDa、3.4–5 kDa和5–10 kDa的胶条分别切下,将胶条均分为1 mm3胶粒,然后进行脱色、干燥。所得胶粒使用胰酶过夜酶解后,抽提肽段,蒸干后置于–80 ℃保存备用。
1.2.3 LC-MS/MS分析
消化肽段样品用毛细管 (75 μm i.d.×15 cm,Beijing SpectraPeaks) 反相低pH分离,毛细管填料为C18 (100 Å,3 μm,Michron Bioresources InC)。分离流速为0.3 μL/min,梯度设置为2%–4% B,6 min;4%–10% B,2 min;10%–25% B,27 min;25%–35% B,20 min;35%–80%,5 min (流动相A,0.1%甲酸和2%乙腈;流动相B,0.1%甲酸和100%乙腈)。洗脱组分经纳升级电喷雾离子源接口喷出,进入LTQ Orbitrap Velos分析。质谱采用一级质谱数据依赖的二级质谱扫描模式 (Data dependent MS/MS scan) 碰撞诱导裂解 (CID) 模式碎裂一级离子。一级质谱扫描范围 (300–1 600),分辨率设置为30 000;自动增益控制 (Automatic gain control,AGC) 为106。依次选取一级信号强度最高的20个离子进行二级碎裂分析,碰撞归一化能量 (NCE) 为35%;AGC为104;最大离子注入时间为30 ms;动态排除为40 s。
1.2.4 数据库搜索与MPs鉴定
Maxquant (version 1.5.6.0) 对质谱产出的数据文件 (.raw) 进行蛋白质数据库搜索。数据库为从UniProt下载的酿酒酵母S288C蛋白序列数据库 (Version: 201711)。搜库参数如下:1) 胰酶特异性酶切;2) 固定修饰为半胱氨酸的烷基化修饰 (+57.021 46 Da);3) 可变修饰为甲硫氨酸氧化修饰(+15.994 92 Da);4) 母离子质量误差20 ppm;5) 子离子质量误差0.5 Da;6) 允许最大漏切位点数目为2个;7) 肽段长度≥7 AA;8) 肽段最大修饰为2种。搜库结果采用目标-诱饵库策略进行过滤,并设定肽段和蛋白质鉴定FDR均小于1%。MPs的筛选标准包括以下几点:1) 至少1条长度≥10 AA的唯一肽段;2) 谱图具有高信噪比且所有b/y离子基本都要能匹配上;3) 谱图主峰被匹配;4) 等重序列过滤,评估是否存在I=L,Q[去酰胺基]=E,GG=N。
2 结果与分析
2.1 酿酒酵母漏检蛋白集中分布于低分子量区
据UniProt数据库记录,酿酒酵母中有707个蛋白质属于漏检蛋白,占总注释蛋白的10.5%,这些漏检蛋白主要分布在低分子量区 (图1A)。酿酒酵母中小蛋白质数为509个,其中漏检蛋白所占比例接近30% (图1B)。除此之外,人、结核分枝杆菌、拟南芥中漏检蛋白在小蛋白质中比例相仿,这些漏检蛋白分布特征暗示我们可以通过小蛋白质特殊富集策略来挖掘漏检蛋白。因此我们采用12% Tricine-SDS-PAGE来分离、富集酿酒酵母小蛋白质。
2.2 富集法鉴定72个小蛋白质
我们共鉴定到697个蛋白质 (1 960个唯一肽段),其中小蛋白质72个,占酵母已注释PE1小蛋白质的35%,其中47个蛋白鉴定到两条及两条以上唯一肽段 (表1)。小蛋白质鉴定数据集中最大类群属于核糖体蛋白家族,共鉴定到15个亚基。其中40S鉴定小蛋白数占该类群注释小蛋白的70%,60S鉴定小蛋白占该类群注释小蛋白的54.5%。小蛋白质鉴定数据集中还鉴定到其他类群蛋白,如细胞色素家族、转运蛋白、ATP合成酶亚基家族、U6 snRNA相关蛋白等。
2.3 鉴定小蛋白质特征
相对高分子量:已鉴定小蛋白质长度主要在61–100 AA间 (图2A)。尽管已注释小蛋白质中1–60 AA蛋白数最多,但实际鉴定数最少,这反映了即使在使用小蛋白质鉴定技术的条件下依然存在着向更长蛋白的偏倚。
高比例亲水性蛋白:与已注释小蛋白质相比,72个鉴定小蛋白质中疏水蛋白比例要低很多,尤其在1–60 AA蛋白类群中疏水比例差别最大 (图2B)。总已注释小蛋白质中,疏水蛋白占43%,1–60 AA中疏水蛋白比例高达52.8%。然而,在我们鉴定小蛋白质中,亲水蛋白占主体,比例高达82%。短的相对于长的蛋白的更强疏水倾向特性是造成小蛋白质鉴定向更长蛋白偏倚的一个重要原因。
细胞分布差异不显著:亚细胞定位分析 (图2C) 显示,鉴定小蛋白质主要定位于细胞膜和细胞质,且二者蛋白数目几乎相当。而已注释小蛋白质中绝大多数都定位于细胞膜,与前面描述的高疏水性特征相一致。传统的蛋白提取通常以完整细胞为研究对象,针对膜蛋白的提取策略可能会提高小蛋白质覆盖度。
弱密码子使用偏性:密码子适应指数 (Codon adaptation index,CAI) 反映编码区同义密码子与密码子最佳使用相符合的程度,取值范围在0–1之间。CAI值越高表明更强密码子使用偏性和更高表达水平[15]。已注释小蛋白质编码基因的CAI值在整体上最低 (图2D),表明小蛋白质的表达水平可能要低于蛋白质组整体表达水平。鉴定小蛋白质比已注释小蛋白质对应基因的CAI值要高,意味着我们鉴定到的小蛋白质是已注释小蛋白质中相对高表达的部分。密码子使用偏性被普遍认为受蛋白编码基因长度的影响,在大肠杆菌等细胞中,密码子使用偏性与蛋白编码基因长度呈显著正相关。Eyre-Walker猜测这是由于进化产生的选择压力以避免翻译过程中错误的引入[16]。基于酵母小蛋白质整体弱密码子使用偏性的特征优化其表达条件可能会促进小蛋白质的表达和鉴定。
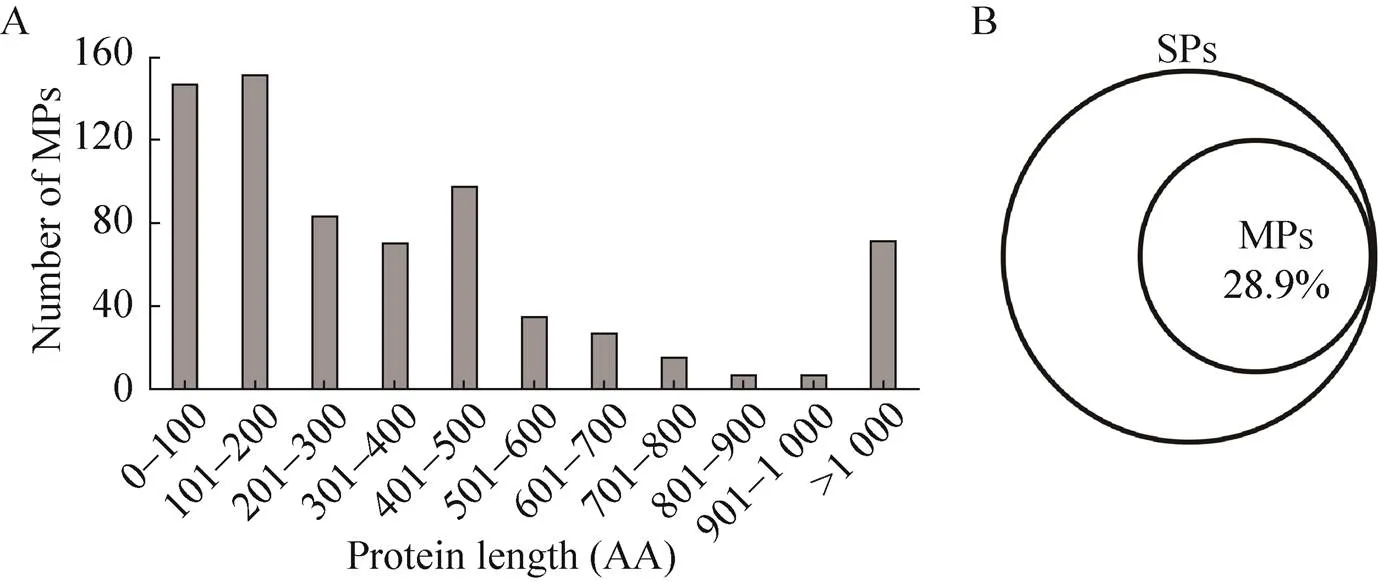
图1 酿酒酵母漏检蛋白分布特征
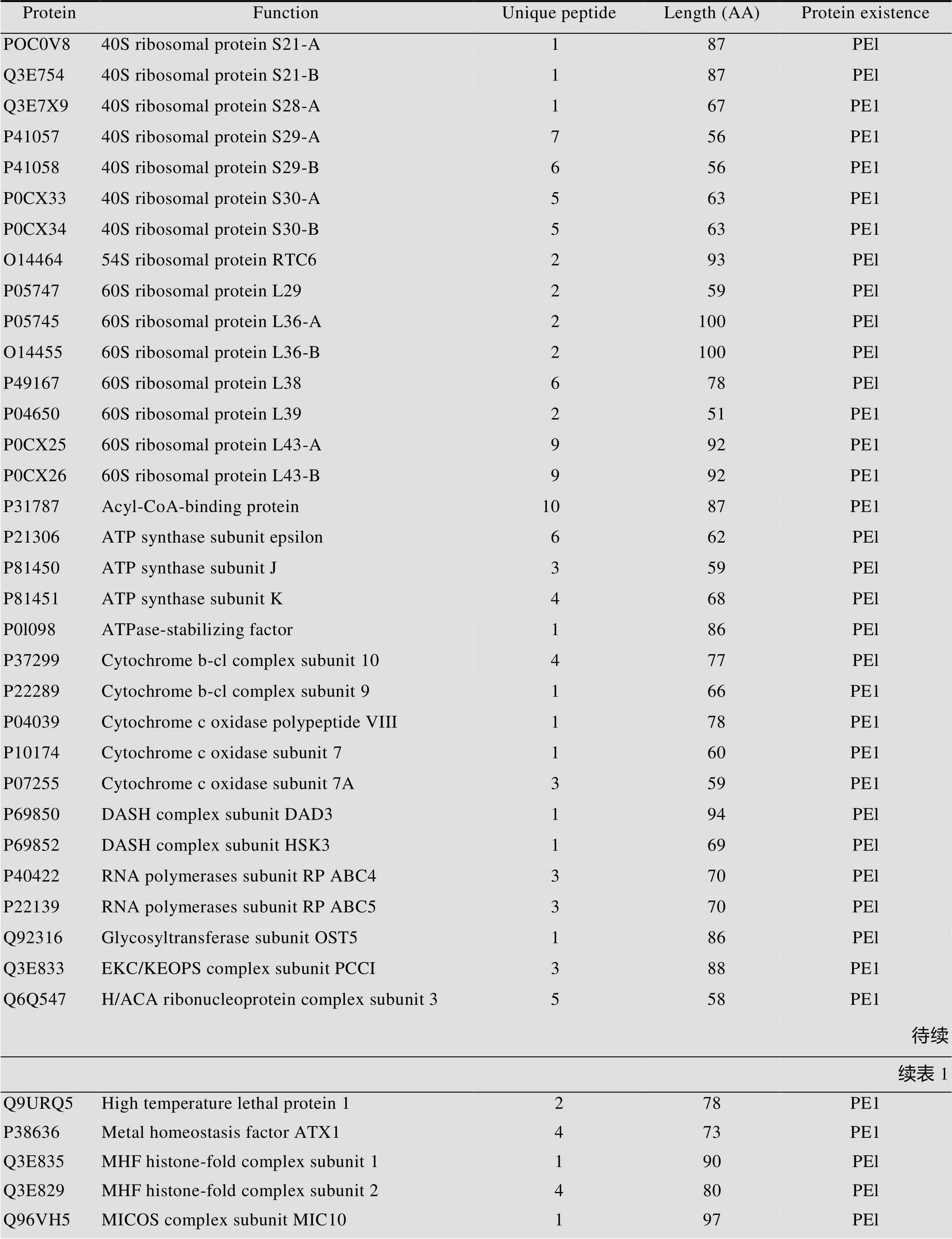
表1 富集法鉴定到72个小蛋白质
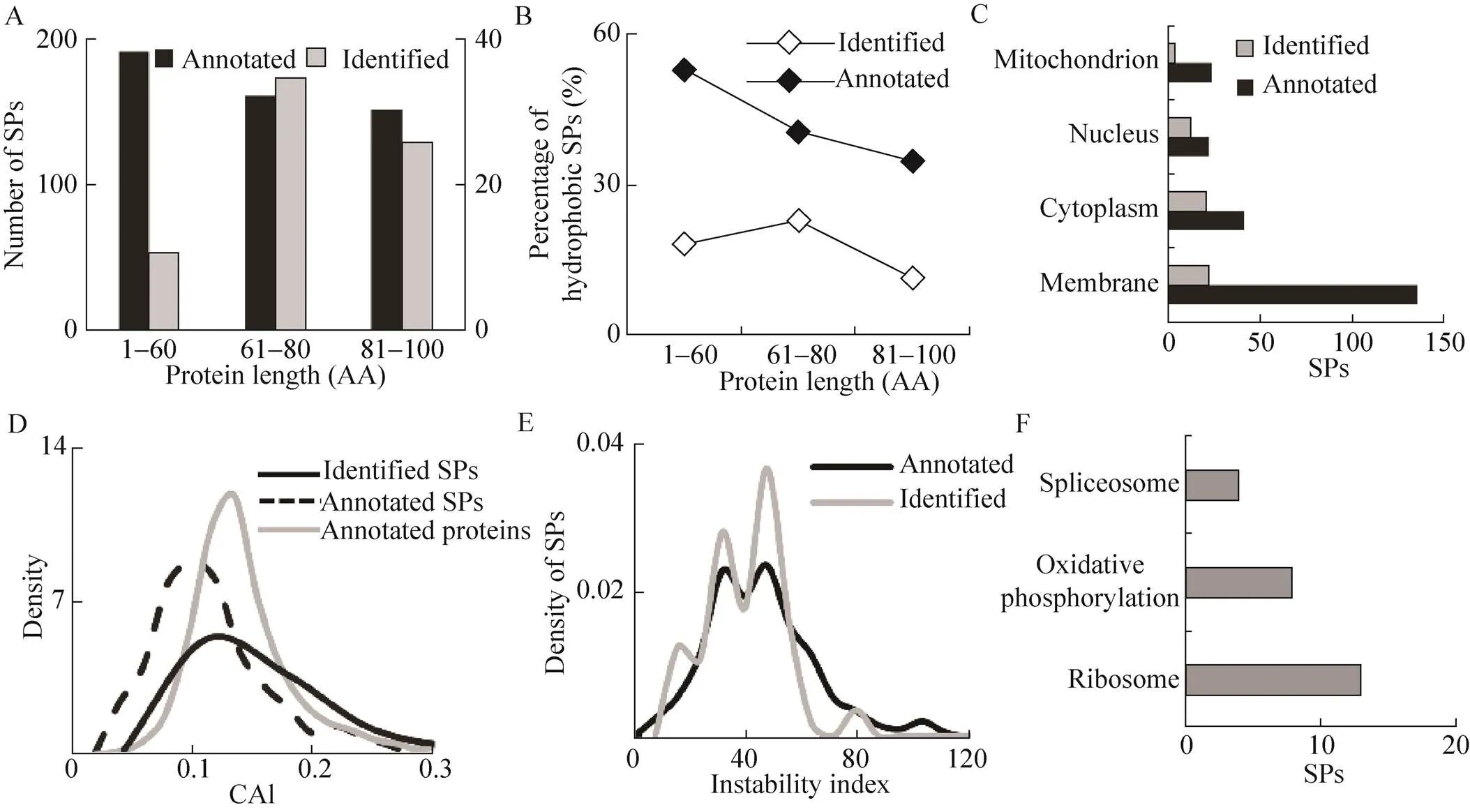
图2 鉴定小蛋白质特征分析
不稳定性:蛋白质不稳定系数 (Instability index) 可用来评估蛋白质稳定性。该参数低于40的蛋白质被认为是稳定蛋白,而高于40则被认为不稳定,不稳定蛋白往往半衰期较短[17]。我们发现一半以上的小蛋白质属于不稳定蛋白质。与已注释小蛋白质相比,鉴定小蛋白质在高稳定性蛋白质区域的比例相对较低且整体呈现向中、低稳定性蛋白质区域的偏倚 (图2E),这意味着不稳定性是小蛋白质的另一重要特征。
KEGG分析 (图2F) 表明已鉴定小蛋白质主要是核糖体和剪接体的组成成分,且主要参与了氧化磷酸化的细胞通路,表明这些小蛋白质在蛋白质翻译和能量代谢方面发挥重要功能。
2.4 小蛋白质富集促进漏检蛋白鉴定
在小蛋白质富集数据集中我们共鉴定到13个漏检蛋白,经严格谱图质量筛选后 (图3E),最终保留了9个漏检蛋白,它们均仅有一条唯一肽段。验证的漏检蛋白中,包括5个PE3 (Q02820、A5Z2X5、Q2V2P4、P38127、P40086) 和4个PE4 (Q2V2P9、Q3E762、Q8TGU7、Q2V2P2) 蛋白质 (图3A)。可信漏检蛋白中7个蛋白质 (Q02820、A5Z2X5、P38127、P40086、Q2V2P9、Q8TGU7、Q8TGU7) 定位于膜上,5个属于单跨膜漏检蛋白,2个线粒体内在膜蛋白质 (P38127和P40086)多次跨膜,分别跨膜6次和7次。这提示我们 可以借鉴膜蛋白富集策略进一步实现MPs高效鉴定。
可信漏检蛋白中,5个漏检蛋白属于功能未知蛋白;Q02820可能参与非经典蛋白输出通路[18];2个线粒体内在膜蛋白可能参与能量转换途径[19],即P38127 (Mitochondrial carrier protein RIM2) 和P40086 (Cytochrome c oxidase assembly protein COX15);A5Z2X5属于UPF0495家族蛋白;Q2V2P4属于功能未知蛋白,位于Ⅸ染色体上,由73 AA组成,其中1–22 AA属于信号肽区,成熟区是23–73 AA区。
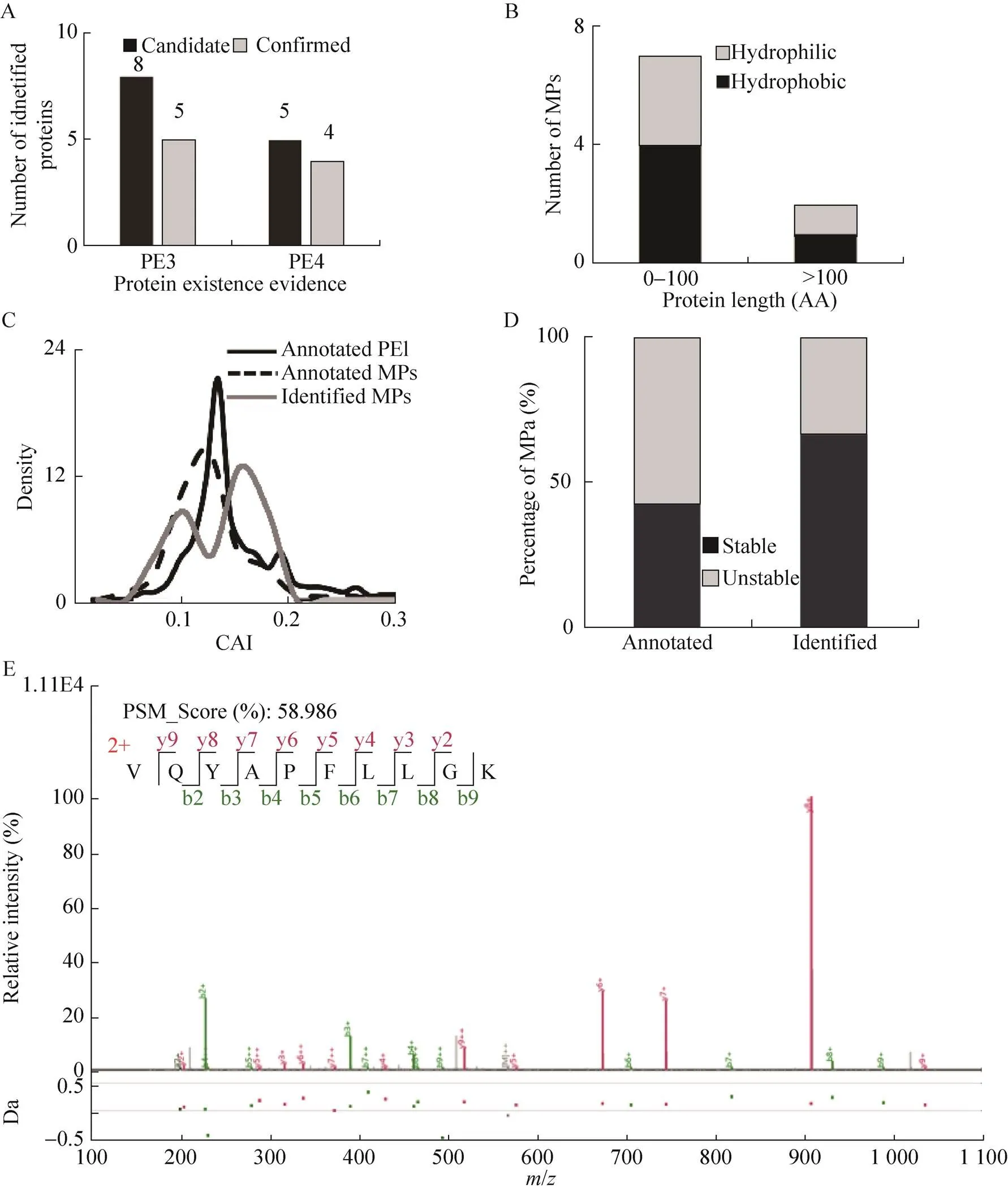
图3 小蛋白质富集促进漏检蛋白鉴定
漏检蛋白中78%的蛋白质属于小蛋白质(图 2B),表明小蛋白质富集策略有助于挖掘漏检蛋白。鉴定漏检蛋白中一半属于亲水蛋白,疏水蛋白鉴定效率的提高可能有助于漏检蛋白的鉴定。
已注释PE1蛋白、已注释漏检蛋白和鉴定漏检蛋白编码基因CAI分布比较分析表明,已注释漏检蛋白对应基因的CAI在整体上要显著低于已注释PE1蛋白,这说明漏检蛋白的表达水平要低于PE1蛋白。此外,与已注释漏检蛋白相比,鉴定漏检蛋白相应基因的CAI在整体上向高CAI区偏移,这表明我们鉴定到的漏检蛋白是已注释漏检蛋白中相对高表达的部分(图 2C)。弱密码子使用偏性可能是造成其相应蛋白难以检测的深层次原因。
已注释漏检蛋白中57%的蛋白属于不稳定蛋白,而在鉴定漏检蛋白中不稳定蛋白仅占33% (图 2D)。这说明高比例不稳定蛋白是漏检蛋白整体的一个重要特征,其可能不利于漏检蛋白的鉴定。提高蛋白稳定性的方法可能有助于进一步鉴定漏检蛋白。
3 讨论
尽管成千上万蛋白质的结构与功能已被系统研究,小蛋白质的生物学研究却被极大地忽视了。这主要是由于小蛋白质的高效鉴定是蛋白质组学领域的技术难题。我们通过富集策略鉴定到72个小蛋白质,占酵母已注释PE1小蛋白质的35%,并成功验证了9个漏检蛋白。一些难检测的小蛋白质和漏检蛋白往往具有类似特征,如低分子量、低丰度、强疏水性、膜结合、弱密码子使用偏性以及蛋白不稳定性等。这些典型的特征暗示我们后续可在以下几方面进行优化:1) 表达条件的优化[20];2) 蛋白质半衰期的延长;3) 基于亚细胞定位的提取策略(如膜蛋白质);4) 消化策略的改进(针对疏水蛋白的多酶组合、化学切割[21]、消化条件[22]以及自上而下蛋白质组学策略[23])。随着对小蛋白质特性的深入理解、样品制备技术的改进以及质谱技术的发展,有望实现小蛋白质的深度覆盖,这将为其功能探究奠定技术支撑。
[1] Zanet J, Benrabah E, Li T, et al. Pri sORF peptides induce selective proteasome-mediated protein processing. Science, 2015, 349(6254): 1356–1358.
[2] Costa LM, Marshall E, Tesfaye M, et al. Central cell-derived peptides regulate early embryo patterning in flowering plants. Science, 2014, 344(6180): 168–172.
[3] Cabrera-Quio LE, Herberg S, Pauli A. Decoding sORF translation-from small proteins to gene regulation. RNA Biol, 2016, 13(11): 1051–1059.
[4] UniProt Consortium. The universal protein resource (UniProt). Nucleic Acids Res, 2007, 35(D1): D193–D197.
[5] Archakov A, Zgoda V, Kopylov A, et al. Chromosome-centric approach to overcoming bottlenecks in the Human Proteome Project. Exp Rev Proteom, 2012, 9(6): 667–676.
[6] Huh WK, Falvo JV, Gerke LC, et al. Global analysis of protein localization in budding yeast. Nature, 2003, 425(6959): 686–691.
[7] Ghaemmaghami S, Huh WK, Bower K, et al. Global analysis of protein expression in yeast. Nature, 2003, 425(6959): 737–741.
[8] Sun YH, de Jong MF, den Hartigh AB, et al. The small protein CydX is required for function of cytochrome bd oxidase in. Front Cell Infect Microbiol, 2012, 2: 47.
[9] Ma J, Diedrich JK, Jungreis I, et al. Improved identification and analysis of small open reading frame encoded polypeptides. Anal Chem, 2016, 88(7): 3967–3975.
[10] Xu P, Duong DM, Seyfried NT, et al. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell, 2009, 137(1): 133–145.
[11] Wu FL, Zhao MZ, Zhang Y, et al. Recombinant acetylated trypsin demonstrates superior stability and higher activity than commercial products in quantitative proteomics studies. Rapid Commun Mass Spectrom, 2016, 30(8): 1059–1066.
[12] Zhao MZ, Wu FL, Xu P. Development of a rapid high-efficiency scalable process for acetylated Sus scrofa cationic trypsin production from Escherichia coli inclusion bodies. Prot Exp Purificat, 2015, 116: 120–126.
[13] Zhai LJ, Chang C, Li N, et al. Systematic research on the pretreatment of peptides for quantitative proteomics using a C18microcolumn. Proteomics, 2013, 13(15): 2229–2237.
[14] Xu P, Duong DM, Peng JM. Systematical optimization of reverse-phase chromatography for shotgun proteomics. J Prot Res, 2009, 8(8): 3944–3950.
[15] Sharp PM, Li WH. The codon Adaptation Index–a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res, 1987, 15(3): 1281–1295.
[16] Eyre-Walker A. Synonymous codon bias is related to gene length in: selection for translational accuracy? Mol Biol Evolut, 1996, 13(6): 864–872.
[17] Wilkins MR, Gasteiger E, Bairoch A, et al. Protein identification and analysis tools in the ExPASy server//Link AJ, ed. 2-D Proteome Analysis Protocols. Totowa: Humana Press, 1999, 112: 531–552.
[18] Cleves AE, Cooper DN, Barondes SH, et al. A new pathway for protein export in Saccharomyces cerevisiae. J Cell Biol, 1996, 133(5): 1017–1026.
[19] Barros MH, Carlson CG, Glerum DM, et al. Involvement of mitochondrial ferredoxin and Cox15p in hydroxylation of heme O. FEBS Lett, 2001, 492(1/2): 133–138.
[20] Quax TE, Claassens NJ, Söll D, et al. Codon bias as a means to fine-tune gene expression. Mol Cell, 2015, 59(2): 149–161.
[21] Fischer F, Poetsch A. Protein cleavage strategies for an improved analysis of the membrane proteome. Proteome Sci, 2006, 4: 2.
[22] Min L, Choe LH, Lee KH. Improved protease digestion conditions for membrane protein detection. Electrophoresis, 2015, 36(15): 1690–1698.
[23] Moradian A, Kalli A, Sweredoski MJ, et al. The top-down, middle-down, and bottom-up mass spectrometry approaches for characterization of histone variants and their post-translational modifications. Proteomics, 2014, 14(4/5): 489–497.
Identification of missing proteins inbysmall protein-based enrichment
Cuitong He1,2, Yao Zhang2,3,Ying Jiang1,2, and Ping Xu1,2
1 Anhui Medical University, Hefei 230032, Anhui, China 2 Beijing Institute of Radiation Medicine, State Key Laboratory of Proteomics, National Center for Protein Sciences (Beijing), Beijing Proteome Research Center, Beijing 102206, China 3 School of Life Sciences, Sun Yat-sen University, Guangzhou 510275, Guangdong, China
Small proteins (SPs) are defined as peptides of 100 amino acids or less encoded by short open reading frames (sORFs). SPs participate in a wide range of functions in cells, including gene regulating, cell signaling and metabolism. However, most annotated SPs in all living organisms are currently lacking expression evidence at the protein level and regarded as missing proteins (MPs). High efficient SPs identification is the prerequisite for their functional study and contribution to MPs searching. In this study, we identified 72 SPs and successfully validated 9 MPs frombased on SPs enrichment strategy. In-depth analysis showed that the missing factors of MPs were low molecular weight, low abundant, hydrophobicity, lower codon usage bias and unstable. The small protein-based enrichment can be used as MPs searching strategy, which might provide the foundation for their further function research.
proteomics, small protein, enrichment, missing protein
January 30, 2018;
March 12, 2018
Chinese National Basic Research Programs (No. 2017YFC0906600), National Natural Science Foundation of China (No. 31670834), Beijing Training Project for The Leading Talents in S&T (No. Z161100004916024).
Ping Xu. Tel: +86-10-61777113; Fax: +86-10-61777050; E-mail: xupingghy@gmail.com
Ying Jiang. Tel: +86-10-80705299; Fax: +86-10-80705002; E-mail: jiangying304@hotmail.com
10.13345/j.cjb.180046
国家重大研发计划精准医学重大专项 (No. 2017YFC0906600),国家自然科学基金(No. 31670834),北京市百名领军人才计划 (No. Z161100004916024) 资助。
(本文责编 郝丽芳)
猜你喜欢
林业调查规划(2021年6期)2022-01-19
生物学通报(2020年11期)2020-10-22
发明与创新·中学生(2019年6期)2019-06-26
中国人兽共患病学报(2018年12期)2019-01-21
中成药(2018年7期)2018-08-04
中老年健康(2018年2期)2018-04-10
中国调味品(2017年2期)2017-03-20
中国调味品(2017年2期)2017-03-20
中国科技信息(2015年2期)2015-11-16
中国卫生统计(2015年4期)2015-03-09
