
α-二氧化锰对钾离子吸附性能的研究及计算
2019-01-16 12:11张冬昊黄雪莉
无机盐工业 2019年1期
张冬昊,黄雪莉,刘 娜
(新疆煤炭洁净转化与化工过程重点实验室,新疆大学化学化工学院,新疆乌鲁木齐830046)
利用吸附的方式,从低浓度海水或盐湖卤水中高效提取钾一直是各国学者研究的重要领域之一[1-2]。M.Kamatsu[3]在专利中提出钛氧化物用于海水中K+的吸附;J.Hou等[4]合成的沸石分子筛对海水中K+的吸附量为 54.9 mg/g;石勤等[5]合成的钡十字沸石分子筛对玛纳斯盐湖卤水中K+吸附量为70.1mg/g,对罗布泊盐湖卤水中K+的吸附量达85.1 mg/g。
类似于多孔结构的沸石分子筛,二氧化锰离子筛作为离子交换中较具优势的一种吸附剂[6],近年来逐渐为学者所关注。 自然界中存在α、β、γ、δ、ε和λ 等多种晶型[7-8]的二氧化锰,主要以[MnO6]正八面体为单元,相邻八面体共角或共棱组成含(1×1)、(1×2)、(2×2)隧道的层状、立体骨架状空间结构,各晶型的MnO2均具有不同的选择吸附作用[9-11]。α-MnO2空间群为 I4/m,有(1×1)和(2×2)空间隧道,其中(2×2)隧道尺寸为 0.46 nm,与一系列水合离子(K+、NH4+、Ba2+等)半径接近[12],且由于晶格缺陷还存在Mn3+造成电荷失衡,这两点为α-MnO2吸附K+提供了理论条件。董殿权等[13]合成出隐钾锰矿型MnO2,并测定了其对碱金属离子的分配系数由大到小依次为 K+、Rb+、Na+,交换性能随 pH 增加而增大,对 K+的饱和交换容量达到181.5 mg/g。刘月[14]对几种二氧化锰晶体和表面结构进行了理论研究,主要涉及铁磁性和反铁磁性的计算。但总的来说,大多数α-MnO2合成方法周期较长,吸附量不高,有必要进一步研究,包括α-MnO2吸附K+过程的模拟研究。
本文以水热法合成α-MnO2离子筛,探究了物料物质的量比、水热温度、晶化时间、反应溶液中酸浓度对样品吸附性能的影响,并基于密度泛函理论采用Material Studio8.0进行了分子模拟计算。
1 实验部分
1.1 实验分析设备及主要药品
采用D8型X射线衍射仪(XRD)对样品的晶体结构进行测试;釆用VERTEX70型红外光谱仪(FTIR)对样品进行测试;采用SU8000型扫描电子显微镜(SEM)观察样品形貌;采用ESCALAB 250Xi型X射线光电子能谱探究样品化合价。
高锰酸钾(KMnO4)、硫酸锰(MnSO4·H2O)、氢氧化钾(KOH)、氯化钾(KCl),均为分析纯;所用水为去离子水。
1.2 α-MnO2离子筛的制备与改性
α-MnO2离子筛合成方法众多,溶胶凝胶法多以有机溶剂作为助剂,污染较大;氧化还原沉淀法和水热合成法条件容易控制,产品晶型较好,操作简单、无污染且反应均匀,但操作时间相对较长。本文选择水热合成法合成α-MnO2离子筛,晶体熟化遵循奥斯瓦尔德熟化机理,生成晶体后在高温高压下溶解再重结晶成稳定晶体。以一定比例的MnSO4和KMnO4为原料,充分溶解搅拌后转移至高温水热釜,恒温条件下静置晶化一定时间,过滤干燥得到样品。
α-MnO2离子筛在含K+溶液中合成,K+在晶体结构的空腔中起支撑作用。通过酸处理,可以在不改变结构的同时以H+替换K+所在的位置,达到改性的目的。因此,本文中α-MnO2离子筛的改性采用酸浸法,以一定浓度硝酸浸泡水热合成的含钾二氧化锰离子筛一定时间,抽滤并反复冲洗至中性后干燥,得 K+被抽出(2×2)隧道的 H 型 α-MnO2离子筛。
1.3 α-MnO2离子筛的吸附
参考海水和盐湖卤水中K+的浓度,配制含K+质量分数为0.5%左右的溶液,准确称取0.2 g吸附剂置入锥形瓶,加入150 mL含钾溶液,恒温水浴振荡吸附足够时间后,测上清液中K+浓度。单位质量α-MnO2对K+吸附量的计算公式为:
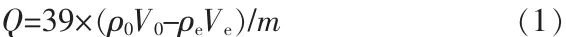
式中,Q表示α-MnO2离子筛对K+的吸附量,mg/g;ρ0、ρe表示初始条件和吸附结束时溶液中K+的质量浓度,g/L;V0、Ve表示初始条件和吸附结束时溶液的体积(体积基本不变),mL;m表示所取吸附剂α-MnO2离子筛质量,g。
2 α-MnO2离子筛的模拟计算研究
2.1 模型构建
采用无机晶体结构数据库(ICSD)提供的α-MnO2离子筛晶胞结构参数和原子位置,空间群为I4/m,晶胞参数为 a=b=0.978 5 nm,c=0.286 3 nm,α=β=γ=90°。 在 Material Studio8.0 软件中,以 2×2×2 超胞作为局域周期性晶体模型,晶胞模型的建立和优化采用CASTEP模块,利用Materials Visualizer建立溶剂模型,采用Forcite模块对模型进行优化。
2.2 模拟方法
为简化计算,本文仅以纯α-MnO2晶体为例进行计算讨论。运用DFT平面波赝势法,进行第一性原理计算。交互相关函数选择PBE/GGA(广义梯度近似函数),为克服广义GGA函数的局限性比如带隙值偏小的问题,库伦校正系数U为2.6 eV。同时考虑到计算的有效性和精确性,k-points设置为fine、截断能为340 eV时几何机构和力的优化过程能很好的收敛,费米能级面扩展中高斯拖尾效应为0.1 eV。吸附过程的性质通过巨正则蒙特卡洛(GCMC)方法模拟,采用Sorption模块,计算任务为Locate,计算方法选择 Metropolis,平衡步数为 1×108,生产步数为1×105,温度为25℃。为适应MnO2周期晶体结构,计算采用周期性边界条件,力场为COMPASSⅡ,用密度泛函理论计算离子筛原子电荷,静电势能和Vander Waals势能分别采用Ewald加和方法和Atom based方法。
3 结果与讨论
3.1 α-MnO2离子筛的表征
对初始样品、改性后样品、吸附后样品进行FT-IR、XRD、SEM和XPS表征后,从晶体结构、形貌及价态等方面综合分析离子筛。一系列样品的结果对比见图1~3。所选样品初始合成条件为:n(K)∶n(Mn)=1∶2、反应温度为 90 ℃、酸浓度为 1 mol/L、反应时间为14 h。
二氧化锰离子筛FT-IR表征结果见图1a。图1a所示结果与文献[15]中α-MnO2晶体谱图吻合,3366、3 373、3 392、1 619、1 621、1 384、1 115、960 cm-1为H2O或H3O+的伸缩振动吸收特征峰,也就是晶体孔道内水,而 708~715 cm-1、518~526 cm-1和 466~470 cm-1为[MnO6]正八面体框架中Mn—O键的伸缩振动吸收峰,与α-MnO2的FT-IR波谱数据一致。如图1a所示,经过改性和吸附改性后样品在1 384、1 115、960 cm-1处的吸收峰消失,推测为改性样品结构中暴露出的—OH与 H+或 K+结合成 H3O+或K+·H2O,以至于样品部分吸收峰消失。样品证明为所需的α-MnO2离子筛,并且在改性后及吸附后样品的晶体固有性质没有改变。
合成样品XRD表征如图1b所示。改性前后与吸附后晶体的出峰位置大致不变,与PDF(JCPDS 44-1404)标准卡片的出峰位置和强度基本吻合,没有其他杂峰出现,证明为四方相I4/m空间群的α-MnO2晶体。经过对XRD表征曲线的计算,单元晶胞为a=b=0.978 91 nm、c=0.285 66 nm,与标准卡片十分匹配。改性后的α-MnO2峰位强度比改性前明显增强且尖锐说明晶体纯度更高,吸附后在(110)晶面和(200)晶面出峰位置处的峰强略有降低且峰变宽,进一步验证晶体在吸附K+之后晶胞尺寸变大。
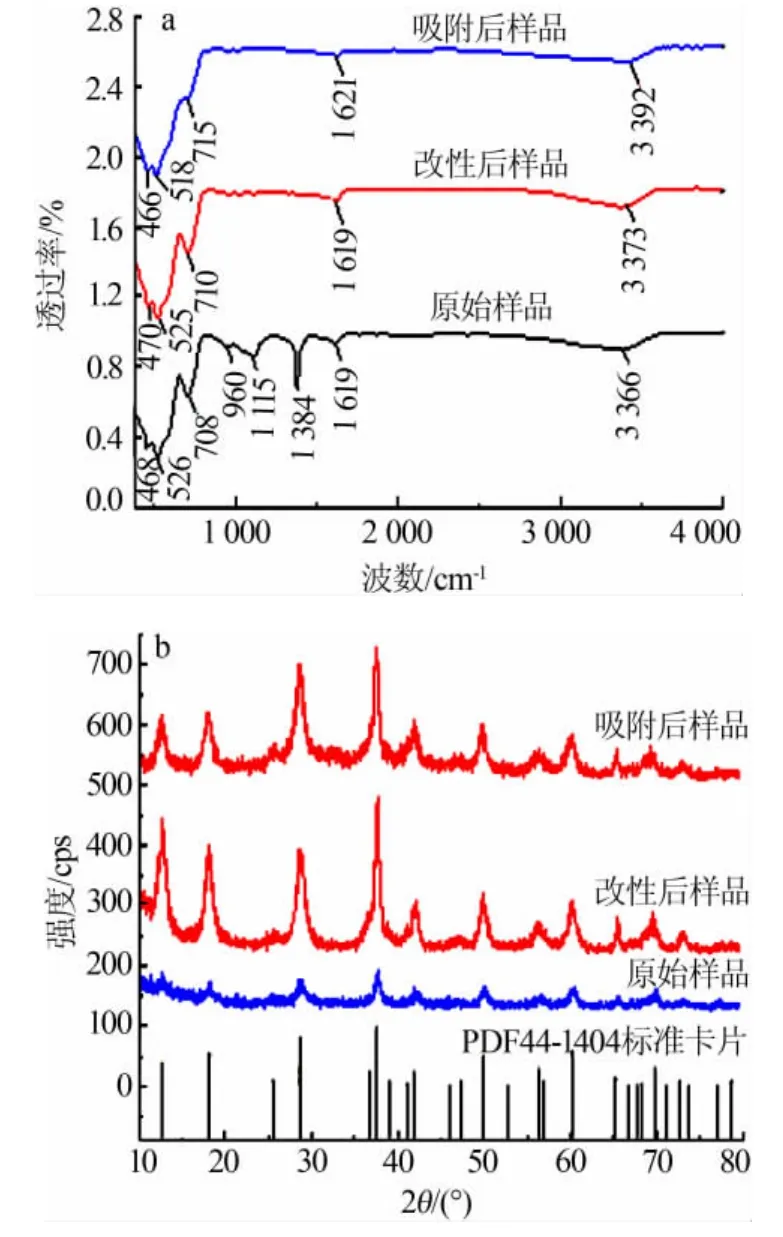
图 1 样品的 FT-IR(a)和 XRD(b)表征结果
对改性后的α-MnO2离子筛进行XPS数据分析,结果如图2所示。图2a为晶体各主元素的分峰,峰面积之比代表相对含量,图2a的Mn与O峰面积之比约为1∶2,证明样品为MnO2。根据MnO2结合能得知,642.7 eV处的Mn2p正是代表了α-MnO2中Mn元素的分峰。同元素不同价态的比较可以通过分析单一元素分峰图得到。图2b为Mn2p分峰图,Mn2p轨道含Mn2p1/2(结合能653.3 eV)和Mn2p3/2(结合能 641.7 eV),分别对应 Mn3+和 Mn4+,通过计算对应 XPS 曲线峰面积可得 n(Mn3+)∶n(Mn4+)约为 1∶1.05。由电荷平衡原理可知,Mn3+的存在使得整体缺正电荷才令离子筛具有阳离子空位,有了吸附K+的电荷基础,根据 n(Mn3+)/n(Mn4+)可计算得本组样品最大吸附量为218.67 mg/g。
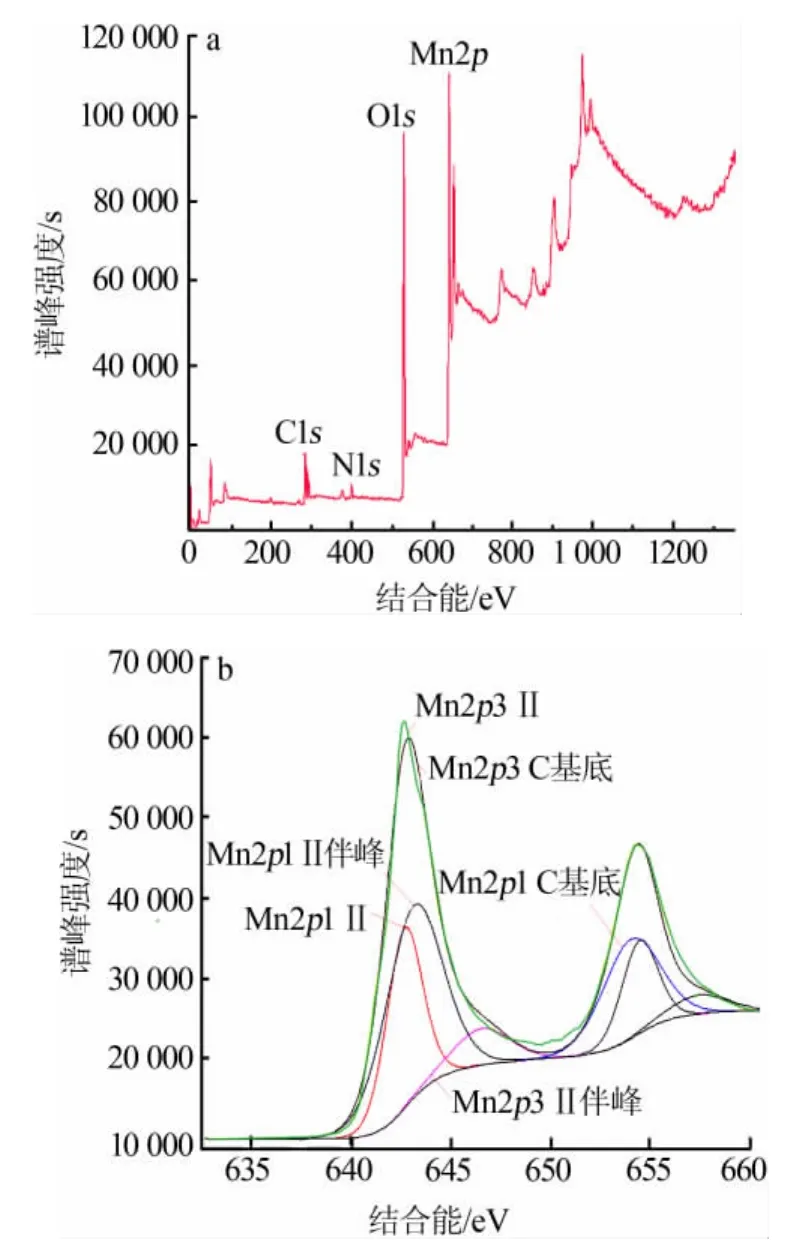
图2 α-MnO2晶体XPS表征数据图
合成样品的SEM形貌如图3所示。由图3可见,合成的晶体形貌结构呈矩形条状或棒状。由于α-MnO2具有各向异性生长的特性,晶体陈化由晶核为引发点沿不同方向生长成为团簇,在水热条件下形成均匀I4/m四方相方形断面的纳米棒,并且纳米棒团聚成花团。但经酸改性,晶体形貌发生一定的改变,花团状团簇被破坏,棒状变为杂乱无章的线状。推测为H+替代K+位置后,单一晶胞尺寸略缩致使整个形貌有区别,原有的团簇被打乱,呈无序小棒状。吸附后较吸附前形貌无明显区别,但分布更为均匀,由改性后的线状略转为矩形条状。

图3 样品SEM图
3.2 α-MnO2离子筛的合成条件对吸附性能的影响
通过单因素实验,研究了合成条件对吸附剂吸附性能的影响,实验结果如图4所示。
从图4可以看出,钾的吸附量随MnSO4用量增加先增加后不变,n(MnSO4)∶n(KMnO4)=3 时最高,为203.57 mg/g;钾吸附量随合成温度升高先增加后减少,110℃时最高,达到214.48 mg/g;钾吸附量随硫酸浓度增大先增加后减少,在硫酸浓度为1.5 mol/L时最高,为225.39 mg/g;钾吸附量在反应时间为12 h时最高,为236.3 mg/g,各组样品最高吸附量均高于文献值 181.54 mg/g[13]。
图4 吸附量随4个因素的变化
吸附完成后,需要对吸附剂中的钾进行脱附,以获得钾的富集液。为了将吸附剂结构中K+抽出,使其能够尽可能转为H型α-MnO2,必须要对样品进行酸处理。经过酸处理后的样品需要用去离子水冲洗,直至洗涤后的上层清液呈中性。酸处理过后以无水乙醇和蒸馏水交替洗涤,不需多次洗涤干燥样品,即可快速达到洗涤目的。被选样品的循环实验结果见图5,在5次吸脱附循环后,样品的吸附量由214.48 mg/g降至181.75 mg/g,然后保持不变,证明该样品有着优异的吸附性能和再生性能。
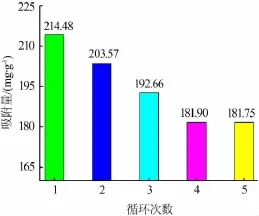
图5 吸附量随循环次数的变化
3.3 α-MnO2离子筛的模拟计算研究
理论计算值空间群为I4/m,晶胞参数为a=b=0.978 5 nm、c=0.286 3 nm,实验结果值空间群为I4/m,晶胞参数为a=b=0.974 93 nm、c=0.283 76 nm,实验值与理论晶胞参数相符。通过对离子筛吸附构型和相应的吸附性能进行计算比较,对吸附能、吸附位及最大理论吸附量等进行分析。计算得吸附能ΔH为-0.030 7 kJ/mol,证明吸附反应为放热过程,因此α-MnO2适合在高温下脱附、低温下吸附。经Sorption模块计算,吸附位为离子筛(2×2)隧道,饱和吸附示意图如图6所示,最大理论吸附量为299.62 mg/g。通过对比计算值和实验值发现,实验测得最大吸附量为计算值的78.87%,仍有较大的提升空间。
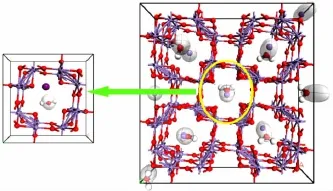
图6 吸附位及饱和吸附示意图
4 结论
通过对 FT-IR、XRD、SEM、XPS 结果分析后得知,制得高纯度纳米棒 α-MnO2[n(Mn3+)∶n(Mn4+)接近1∶1.05]的最优合成条件为:n(MnSO4)∶n(KMnO4)=3、反应温度为110℃、硫酸浓度为1.5 mol/L,反应时间为12 h。样品有着较好的再生性能和吸附性能,最高吸附量为236.3 mg/g,是最大理论吸附量的78.87%。吸附位为离子筛(2×2)隧道,吸附反应为放热过程,低温更适合吸附过程的进行。
猜你喜欢
化工管理(2022年13期)2022-12-02
腐植酸(2021年2期)2021-12-04
人工晶体学报(2021年10期)2021-11-26
粉末冶金技术(2021年3期)2021-07-28
能源工程(2021年1期)2021-04-13
粉末冶金技术(2021年1期)2021-03-29
兰州交通大学学报(2020年5期)2020-11-25
World Journal of Diabetes(2019年7期)2019-07-23
当代陕西(2018年9期)2018-08-29
