
透明质酸基普鲁兰糖载药微球制备与体内外评价
2020-01-15 01:16杨文智赵亚非吴桐刘娇艳李海鹰
河北大学学报(自然科学版) 2020年1期
杨文智,赵亚非,吴桐,刘娇艳,李海鹰
(河北大学 药学院,河北 保定 071002)
微球制剂作为缓控释给药新剂型之一,具有缓释药物、提高药物稳定性、保证疗效、减少给药频次和降低毒副反应等优点,受到药物研究者的青睐[1-2],常见的载药微球可选择天然高分子、合成与半合成的高分子为载体,如:明胶、壳聚糖、淀粉、白蛋白、葡聚糖、海藻酸和透明质酸(HA)等天然高分子;聚乳酸、丙交酯乙交酯共聚物、聚3-羟基丁酸等聚酯类、聚丙烯酸树脂、聚酰胺、聚乙烯醇、乙基纤维素、纤维醋法酯等合成与半合成高分子材料. 普鲁兰多糖(Pu)由α-1,6糖苷键连接的麦芽三糖单元组成,出芽短梗霉分泌,可被淀粉酶降解,具有水溶性、无毒、无致突变作用、无免疫原性、无致畸作用、良好生物相溶性及可生物降解等优良特性,常用作药物载体的材料[3-6]. 为了拓展Pu在医药领域的应用,化学修饰Pu成为研究热点[7-8]. HA同样具有良好的生物相溶性、文献报道HA微球可靶向富含CD44受体的肠癌、乳腺癌和非小细胞肺癌等肿瘤细胞,故被广泛用于缓控释药物的载体[9-11]. 如:与游离阿霉素(DOX)相比,负载DOX的透明质酸-聚γ-苄基-L-谷氨酸胃蛋白体可靶向MCF-7肿瘤细胞,对乳腺癌有更好抑制作用[12];采用HA包载拉帕替尼纳米晶,可抑制肿瘤生长,提高动物存活率[13]. 但单独使用HA作为药物微球载体,需克服其体内迅速降解的缺点[14-16]. 本课题组将HA接枝到Pu糖长链上,制备透明质酸接枝普鲁兰糖(HA-Pu)材料,该新型材料具有较好的生物安全性和抗酶降解性能,可减缓其在体内降解速度[17].本文拟采用抗酶降解的HA-Pu为微球载体材料,经化学交联制备负载抗肿瘤药物阿霉素的HA-Pu微球(DOX-HA-Pu MPs),希望获得具备体内缓释抗瘤的载药微球制剂.
1 实验部分
1.1 仪器、试药与实验动物
LGJ-18冷冻干燥机(宁波新芝仪器有限公司);DVM 6光学显微镜(德国徕卡公司);FTIR-8400s傅里叶变换红外分光光度计(日本岛津仪器公司);T6型紫外可见分光光度计(北京普析通用仪器有限公司);LC 3000型高效液相色谱仪(北京创新通恒科技有限公司).
透明质酸(5400 Da,山东福瑞达医药公司);普鲁兰糖(2000 KDa,日本林原化学株式会社);阿霉素(大连美仑生物技术有限公司提供);透明质酸接枝普鲁兰糖(HA-Pu)材料源于实验室自制[17];其余试剂均是分析纯.
雄性Wistar大鼠(150~200 g),由河北省实验动物中心提供(合格证号:1507012).
1.2 实验方法
1.2.1 HA-Pu微球或载药微球(DOX-HA-Pu MPs)的制备、优化及表征
称取HA-Pu适量,加入蒸馏水搅拌溶解,如:制备载药微球,将DOX加入HA-Pu获得药物与载体材料的水溶液. 将液体石蜡及适量Span 80充分搅拌混匀后,在其中缓慢滴入HA-Pu或载药载体材料水溶液,调节转速,乳化30 min,滴加适量戊二醛液,室温反应1 h,加入异丙醇,搅拌10 min,静置,过滤,乙醚洗涤,干燥,即得HA-Pu MPs或DOX-HA-Pu MPs.
采用BBD实验设计方法优化HA-Pu MPs处方,考察影响微球平均粒径的转速(X1,r/min)、油水比(X2,体积比)和HA-Pu溶液质量浓度(X3mg/mL)3个因素,采用丹东百特 BT-9300ST仪器,测定微球粒径,以微球的平均粒径(Y)为评价指标,建立数学模型,制备17批微球,优化微球处方. 3因素3水平的BBD实验设计见表1.
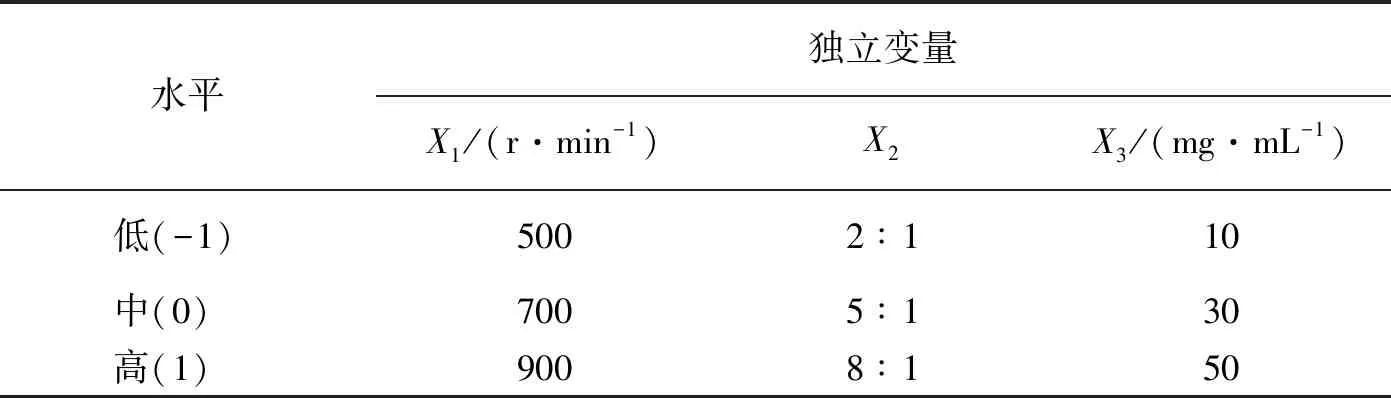
表1 BBD实验因素水平表(n=3)
取冻干后样品于无水乙醇中分散,将微球置于载玻片上,用显微镜观察其微观形态并拍照记录. 样品在红外灯照射下烘干,以干燥的KBr压片,分别测定HA、Pu、HA-Pu、HA与Pu的物理混合物、HA-Pu微球的红外光谱图,扫描波数4 000~400 cm-1,分辨率1 cm-1.
1.2.2 载药微球体外释放
配制盐酸阿霉素标准品液和HA-Pu液,在190~500 nm处紫外扫描,药物紫外最大吸收波长为254 nm,HA-Pu水溶液无干扰. 精密移取200 μg/mL盐酸阿霉素标准液,配制4、6、8、10、12、14和16 μg/mL阿霉素溶液,于254 nm处测定吸光度,以吸收值对质量浓度进行线性回归,获得阿霉素水溶液的标准曲线,考察阿霉素溶液样品日内和日间稳定性.
依照BBD筛选最佳处方,滴加0.5 mol戊二醛,评价药载比(DOX∶HA-Pu,质量比)为1∶10、2∶10、2.5∶10、3∶10、4∶10的载药微球载药量与包封率,获得最佳药载比,制备载药微球,备用. 在透析袋中放入1 mg/mL微球2 mL 混悬液,透析袋置于30 mL pH分别为1.2、6.8和7.4释放液中,在(37±1 )℃,100 r/min恒温振荡,定时吸取透析外液8 mL,同时补充等温等量缓冲液,254 nm处测定紫外吸收度,计算药物质量浓度和药物累计释放量.
取2 mg DOX-HA-Pu载药微球置于5 mL蒸馏水中,超声,4 000 r/min离心10 min,取上清液于254 nm处测定吸光度,依公式(1)和(2)分别计算微球的载药量(DD)与包封率(EE).

(1)
(2)
式(1)、(2)中,M为制得DOX-HA-Pu微球的总质量;M0为受试DOX-HA-Pu微球的质量;M1为受试DOX-HA-Pu微球中DOX的测定值;M2为制备DOX-HA-Pu微球时DOX的投药总量.
1.2.3 药物在大鼠体内药代动力学的测定
用肝素处理过的EP管收集大鼠眼眶血,5 000 r/min的条件下离心10 min,取血浆,备用. 配制0.2 mg/mL的阿霉素标准储备液,置于4 ℃冰箱,避光保存. 取阿霉素储备液,用甲醇配1、5、10、20和40 μg/mL对照品液,分别取10 μL,加入空白血浆190 μL,涡旋混合,加三氯甲烷-甲醇(体积比4∶1)0.5 mL,涡旋混合,在8 000 r/min的条件下离心10 min,取上清液,0.22 μm滤膜过滤,50 ℃氮气吹干,100 μL甲醇复溶,在10 000 r/min离心10 min,取20 μL进样,以峰面积(Y)对血浆药物质量浓度(X,μg/mL)进行线性回归,标准曲线方程为:Y=3 887.5X-1 114.8,R2=0.995(n=3),在1~40 μg/mL内线性关系良好. 取健康雄性Wistar大鼠10只,随机分为2组,禁食12 h,自由饮水,分为腹腔注射10 mg/kg DOX-HA-Pu 微球混悬液组和尾静脉注射等量DOX药物溶液组,给药后0.25、0.5、1、2、4、6、8、12、24、36和48 h眼眶取血,离心,-20 ℃冻存,按照建立标准曲线的方法处理样品,采用HPLC法测定药物质量浓度,绘制药时曲线.
2 结果与讨论
2.1 HA-Pu微球制备
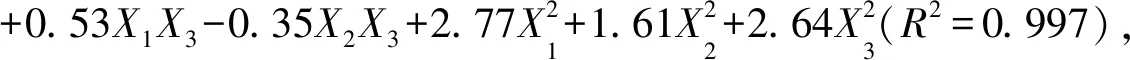
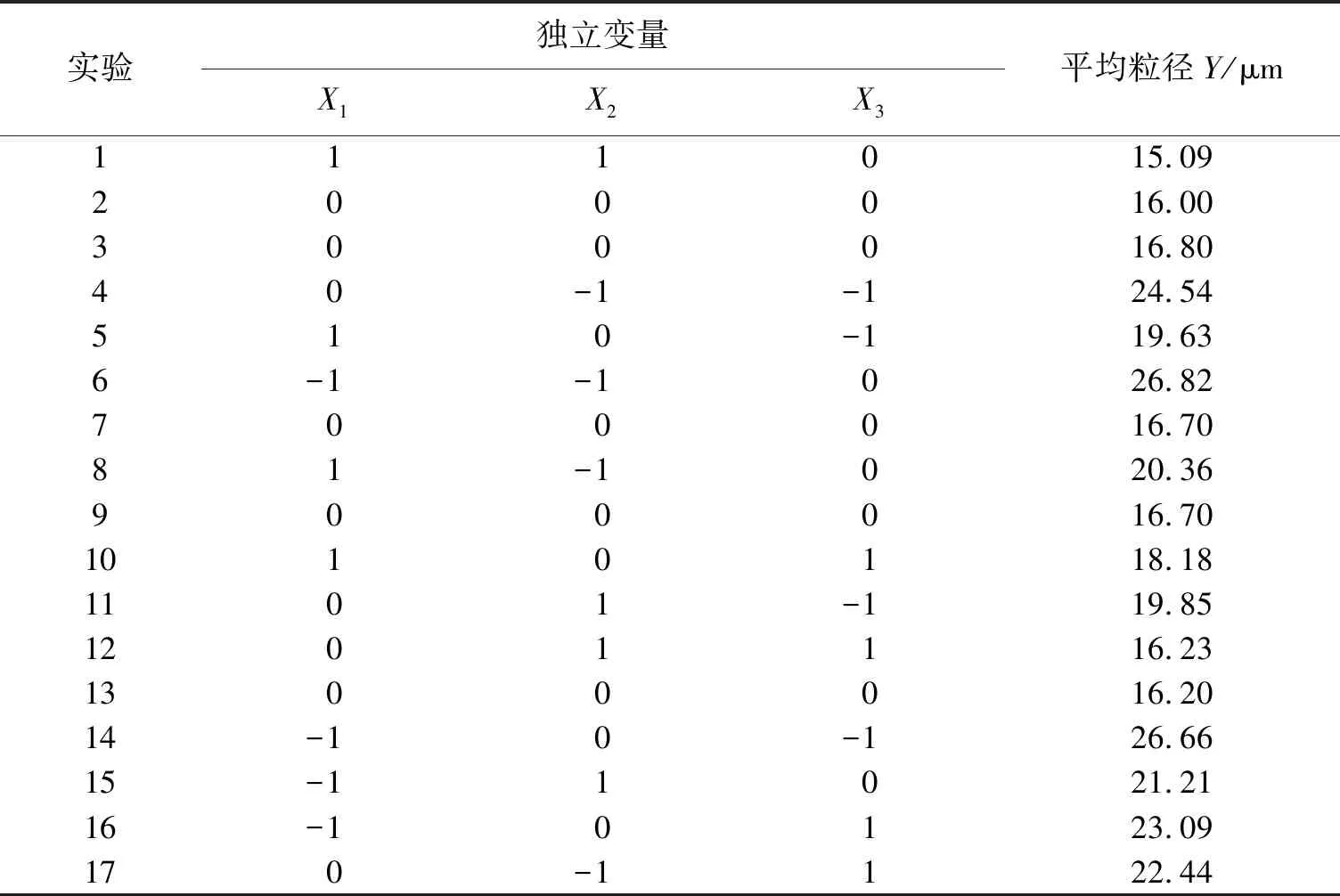
表2 制备微球平均粒径(n=3)
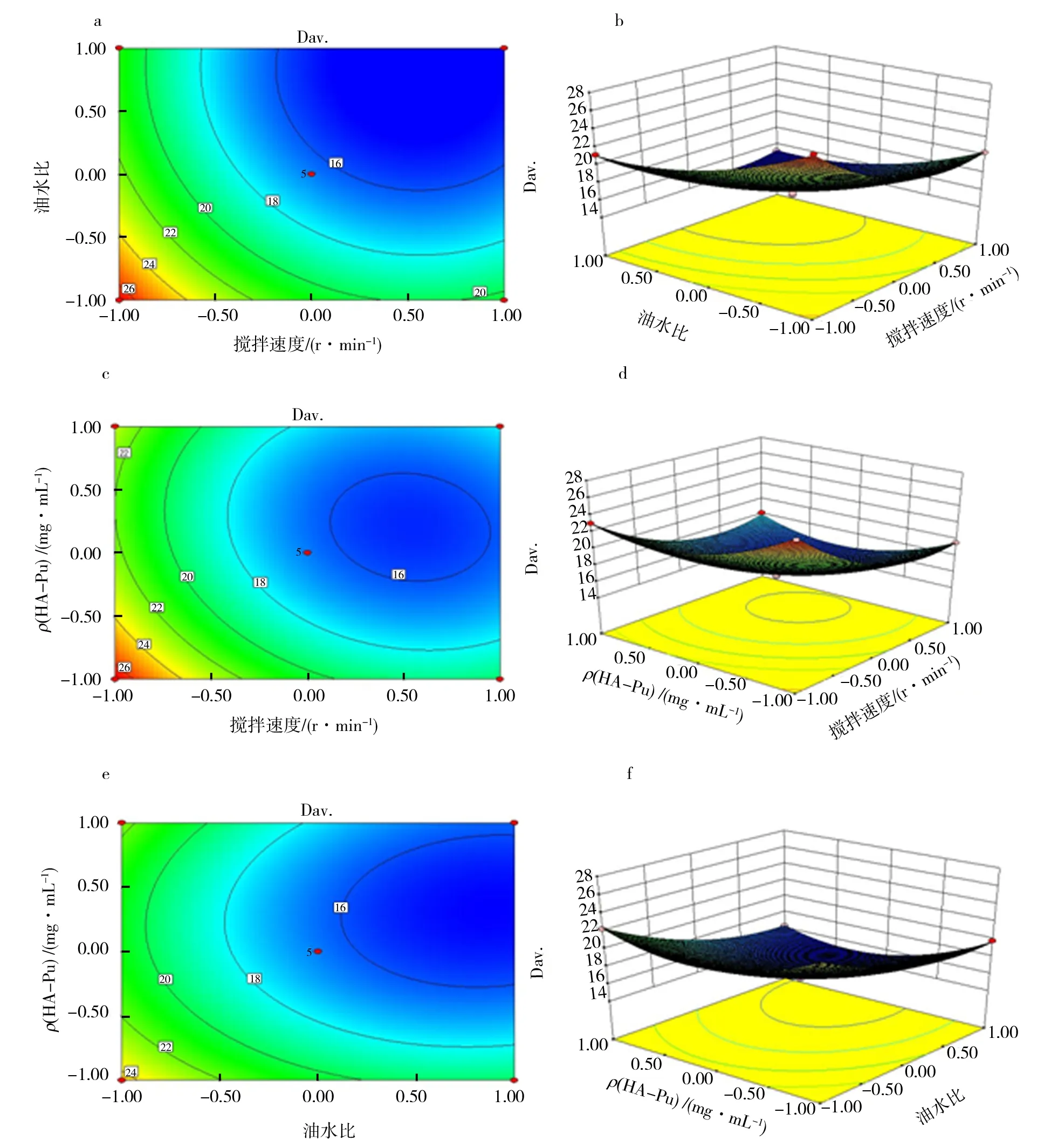
图1 搅拌速度(X1)、油水相比(X2)和 HA-Pu溶液浓度(X3)影响的等值线和曲面
2.2 HA-Pu微球的表征
冻干微球样品的光学显微镜图,见图2,微球球形圆整,表面光滑.红外图谱见图3,HA(3a)、Pu(3b)、HA与Pu物理混合物(3c)、HA-Pu(3d)和HA-Pu 微球(3e)中存在大量—OH与—CH2—基团,3 200~3 600 cm-1显示强的-OH伸缩振动峰,2 925 cm-1出现强的—H伸缩振动峰;HA、HA-Pu和HA-Pu微球中存在—NHCOCH3,故1 655 cm-1出现酰胺Ⅰ带吸收峰,而Pu不含酰胺基团,在1 655 cm-1处未见吸收峰,HA与Pu物理混合物此波数的酰胺吸收峰不明显;HA-Pu 微球相比HA-Pu 材料,在2 850 cm-1处出现游离醛基峰,1 733 cm-1处出现新羰基峰,而1 200~1 000 cm-1缩醛的C—O—C—O—C吸收峰增强,表明HA-Pu与戊二醛成功交联形成微球[21],制备交联微球在光镜下显示出较好形态,见图2.
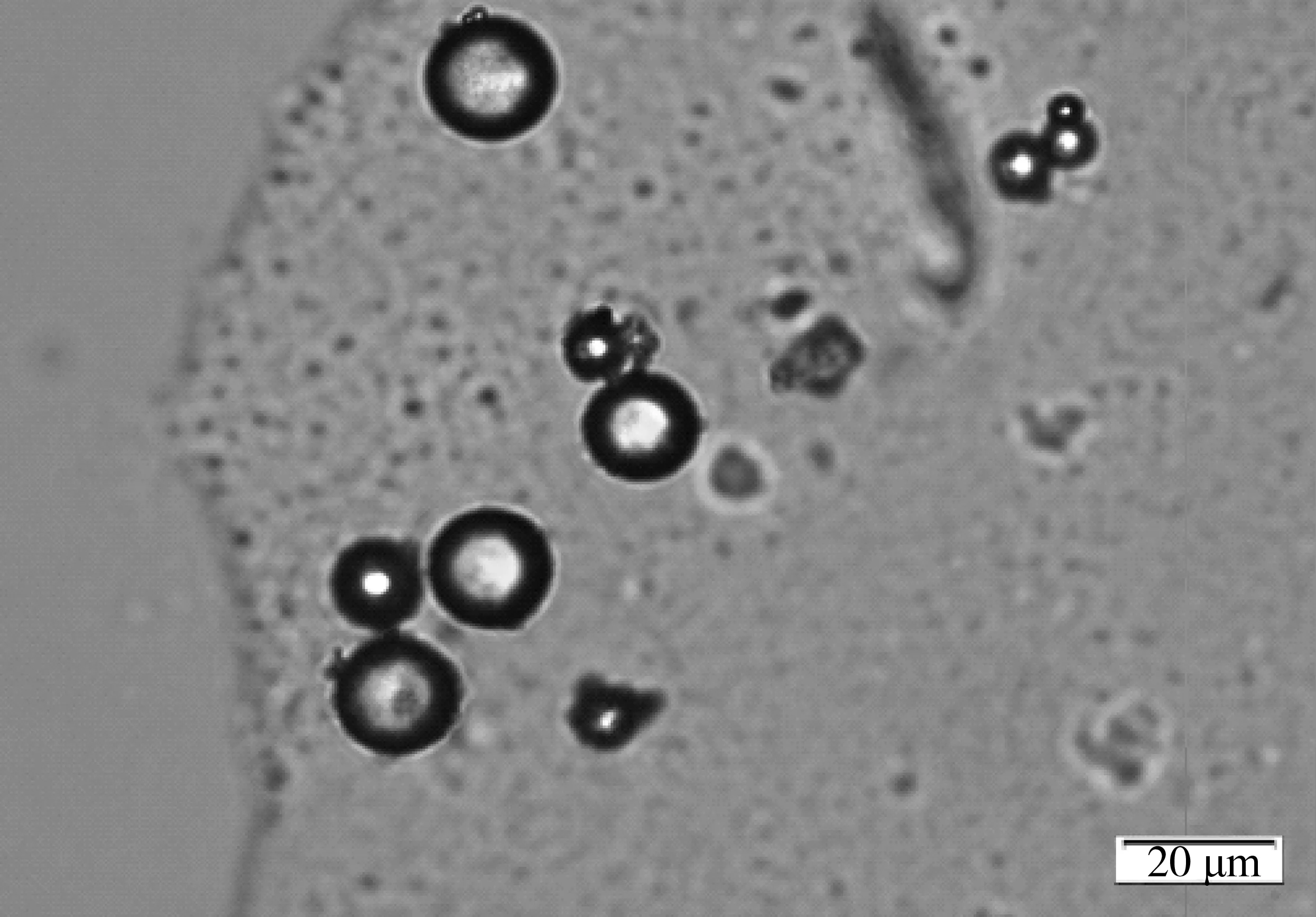
图2 透明质酸接枝普鲁兰糖微球光学显微镜图
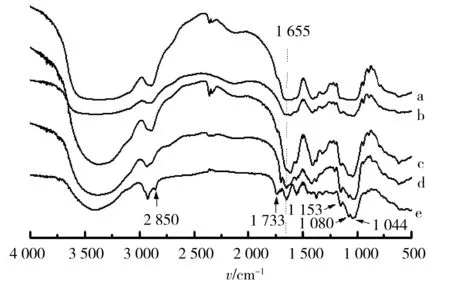
a.透明质酸;b. 普鲁兰糖;c. 透明质酸普鲁兰糖混合物;d.透明质酸-普鲁兰糖;e.透明质酸-普鲁兰糖微球.
2.3 HA-Pu载药微球制备
盐酸阿霉素和HA-Pu水溶液的紫外扫描图显示,盐酸阿霉素最大吸收波长在254 nm,HA-Pu溶液不干扰阿霉素的紫外吸收,测定阿霉素溶液标准方程为y=0.0494x+0.0011(R2=0.999),检测在4~16 μg/mL,阿霉素样品的日内和日间精密度RSD均小于2%. 采用BBD优化条件,制备DOX-HA-Pu MPs,分别采用0.167、0.334、0.5和0.667 mol戊二醛交联微球,考察微球载药量. 结果显示0.5 mol的戊二醛交联,微球获得5.02%最佳载药量. 表3考察不同药载比对HA-Pu MPs的载药量和包封率,DOX与HA-Pu投料比为2∶10(质量比),DOX-HA-Pu MPs获得最佳载药量和包封率.
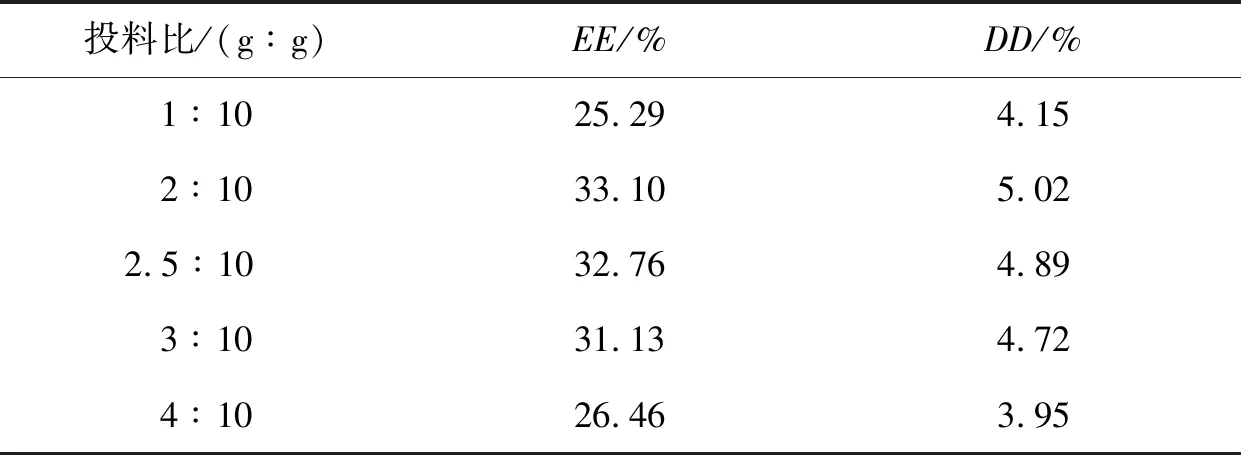
表3 DOX-HA-Pu MPs包封率(EE)和载药量( DD)
2.4 DOX-HA-Pu MPs体外释放
依最优条件制备DOX-HA-Pu MPs,其在不同pH释放介质的释药曲线见图4a.DOX-HA-Pu MPs 前4 h释药,在pH 6.8和7.4释放介质中分别达66%和64%,pH 1.2释药高达87%;24 h时pH 6.8和7.4释放介质释药均接近95%,显示出一定缓释行为. DOX-HA-Pu MPs最初4 h 内释放快,原因是吸附在微球表面和空隙内药物释放导致,而后微球内部的药物通过微球骨架缓慢扩散或HA-Pu材料降解释放,曲线趋于平缓. 将DOX-HA-Pu MPs的体外释药行为拟合,见图4b,拟合度和释放模型拟合结果见表4,由表4可知,DOX-HA-Pu MPs体外释药符合Ritger-Peppas方程,对于圆球形制剂,可根据Ritger-Peppas方程中t的指数n来推测药物从微球骨架结构中释放机制,计算n=0.20≤0.43,故药物的释放以Fick’s扩散方式为主.
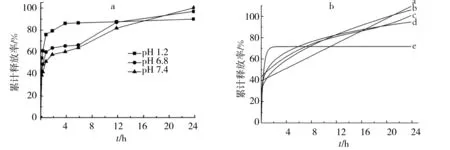
图4 不同pH释放介质下DOX-HA-Pu MPs体外释药曲线(a);pH 7.4释放液的释放模型拟合曲线(b)

表4 DOX-HA-Pu MPs不同模型的释药相关系数(R2)
2.5 载药微球体内药代
测得HPLC条件下盐酸阿霉素大鼠体内的标准曲线方程:A=3 887.514x-1 114.813,R2=0.995(n=5),在1~40 μg/mL内线性关系良好. 4 ℃放置5 d,样品日内日间精密度均符合要求. 图5为Wistar大鼠尾静脉注射DOX·HCl生理盐水溶液和腹腔注射DOX-HA-Pu MPs微球液后的药-时曲线,游离药物注射达峰后迅速下降,24 h难检测,半衰期短(t1/2,4.55 h),代谢较快,与文献报道的3.78 h一致[22]. 而腹腔注射DOX-HA-Pu MPs,t1/2延长至28.64 h,是游离药物的6.3倍;体内平均滞留时间(MRT)为16.77 h,是游离药物的3.5倍,载药微球组药物清除率低. 采用DAS 2.0软件处理药代参数,主要结果见表5.
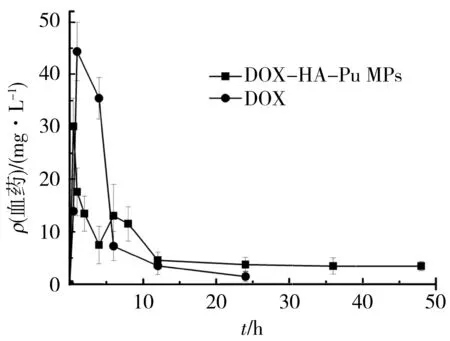
图5 DOX·HCl和DOX-HA-Pu MPs的Wistar大鼠体内药-时曲线 (n=5)

表5 DOX·HCl和DOX-HA-Pu MPs在大鼠体内药代动力学参数(n=5)
T代表受试载药微球制剂,iv表示静脉注射DOX,D表示给药剂量,得到载药微球制剂的腹腔注射的绝对生物利用度为89.6%.
3 结论
采用乳化交联和BBD法,优化HA-Pu MPs制备处方,制备微球表面光滑,形态圆整且粒径均匀,红外谱图显示,成功获得交联微球. 以DOX·HCl为模型药物,制备载药量为5.02%的药物微球制剂. 载药微球体外释放受介质pH影响,偏酸介质可加速微球释药,pH 7.4模拟体液释放环境下,载药微球释药符合Ritger-Peppas方程,其通过Fick's扩散机制释药. 对比大鼠尾静脉注射DOX·HCl与腹腔注射DOX-HA-Pu MPs的药代动力学参数,载药微球释放具备较好的缓释能力.
猜你喜欢
天然产物研究与开发(2022年1期)2022-11-27
中国临床医学影像杂志(2022年6期)2022-07-26
纺织科学研究(2021年6期)2021-12-02
实用皮肤病学杂志(2020年6期)2021-01-27
中国药学药品知识仓库(2021年17期)2021-01-11
中医药学报(2021年9期)2021-01-04
肿瘤防治研究(2019年7期)2019-08-01
中国现代医生(2018年30期)2018-12-06
中国化妆品(2016年4期)2016-11-19
创新时代(2016年4期)2016-05-17
