
美国非处方药专论制度介绍及对我国的启示
2020-04-20 11:09张雨菲董晨东王子哲茅宁莹
中国药房 2020年7期
张雨菲 董晨东 王子哲 茅宁莹
摘 要 目的:为我国实施非处方药(OTC)专论提供参考。方法:采用文献研究法对美国OTC专论的概念、产生与发展历程以及作用等进行全面梳理;借鉴美国经验,探究专论制度对于OTC注册审评的必要性及在当前我国的可实施性,并提出我国实施OTC专论的几点建议。 结果与结论:美国OTC专论是指在销售不包含在新药申请中的OTC产品时应遵循的监管标准,其源于OTC有效性再评价工程,在加速药品上市、促进产品创新方面发挥了特殊作用;OTC专论制度对于优化OTC注册审评具有必要性,但是目前我国尚不具备建立和实施OTC专论的条件,尚需在优化资源配置和基础设施、结合本国OTC管理经验和特点的基础上,才能充分发挥OTC专论在OTC注册审评中的作用,具体可从开展对药品专论深入研究、试点检验专论成效、完善风险管理体系、优化审评资源配置等方面推进。
关键词 非处方药;注册审评;专论制度;美国;启示
ABSTRACT OBJECTIVE: To put forward some suggestions for the implementation of OTC monograph in China. METHODS: Literature research was used to comprehensively review the concept, emergence and development, function of OTC monograph in USA; referring to experience in USA, the necessity and feasibility of monograph system for OTC registration and evaluation in China were explored, and some suggestions were put forward to the implementation of OTC monograph in China. RESULTS & CONCLUSIONS: OTC monograph in USA refers to the regulatory standards that should be followed when selling OTC products not included in new drug applications. The monograph originates from the re-evaluation project of the effectiveness of OTC drugs, which plays a special role in accelerating the drug marketing and promoting product innovation. The OTC monograph system is necessary to help to optimize the OTC registration and evaluation, but at present, the conditions to establish and implement OTC monograph are not yet ripe in China. Based on the optimization of resource allocation and infrastructure construction, national OTC drug management experience, OTC monograph can play a full part in OTC registration and evaluation, in terms of carrying out in-depth research, taking pilot test, improving risk management system and enhancing drug review resource allocation ability.
KEYWORDS OTC drug; Registration and evaluation; Monograph system; USA; Enlightenment
我国自2000年开始实施处方药、非处方药(OTC)分类管理办法,主要对药品分发、销售和使用,即药品上市后的流通环节进行集中管理[1]。但对于上市前的注册准入环节,并没有根据OTC特点设置单独的准入门槛,而是采用与处方药相同的审批流程和技术要求体系。因此,不管是首次上市的新药,或是改变已有药品成分、剂型、工艺等的改良型新药,又或仿制药品,均需要经过严格的临床试验/生物等效性试验、复杂的注册流程及长时间的审批程序,花费3~5年甚至更长时间才能上市,上市速度与处方药品无异,这也使得我国成为世界上OTC注册最为严格的国家之一[2]。过于严苛的技术标准和过于漫长的审批时间与OTC已经通过广泛使用得到充分验证的安全性和有效性并不匹配[3],容易挫傷企业生产积极性,从长远来看不利于我国OTC行业健康发展。目前,我国药品监管部门已认识到分类注册的重要性,并在新修订的《中华人民共和国药品管理法》第十六条中明确规定了优化OTC注册流程,国家药品监督管理局药品审评中心需要根据OTC的特点,制定OTC上市注册相关技术指导原则和程序。尽管具体的OTC注册申请指导方案尚未公布,但对于如何优化管理、提升OTC注册审评效率问题也值得思考。
本文采用文献研究法,通过查阅美国FDA网站上的相关政策,对美国OTC专论(OTC monograph)的概念、产生与发展历程以及作用进行全面梳理。通过分析美国OTC专论的特点,进一步探究当前我国在OTC注册中引入OTC专论的必要性和可行性,并最终为我国实施OTC专论提出几点建议。
1 美国OTC专论的建立与发展历程
1.1 OTC专论的概念
OTC专论是指在销售不包含在新药申请中的OTC产品时应遵循的监管标准[4]。其建立是基于长期以来OTC临床使用经验和使用效果的系统化总结,当一个产品符合专论的全部要求时,即可被认为是安全有效的。
1.2 美国OTC专论的产生与发展历程
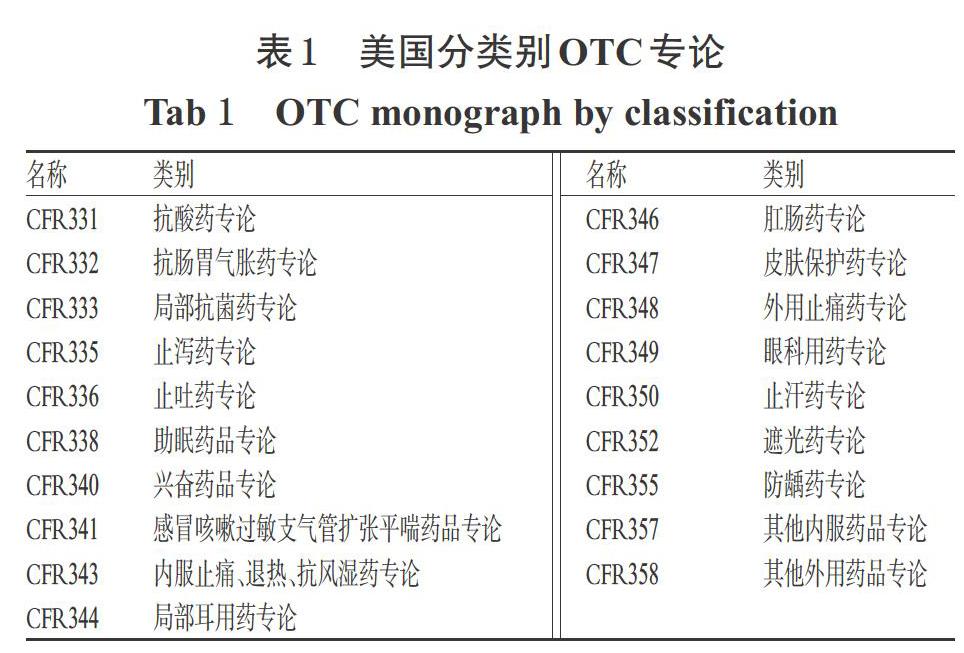
美国在1938年颁布的《食品、药品和化妆品法》中,首次规定“所有新药在上市前必须向美国FDA提供安全性证明”,那时安全性尚作为判断药品是否合规的唯一标准。1962年,美国通过了《药品修正法》(Drug Amendment of 1962),该法规定上市药品不仅要证明其安全性,还要证明其有效性[5]。自此,安全有效便成为药品审评的双项标准。在此背景下,FDA有必要对1962年以前上市的所有药品进行有效性的再评价工作。由于美国在1951年的《Humphrey-Durham修正案》中已正式将处方药和OTC分开,因此在基本完成处方药的有效性再评价之后,1972年FDA药品审评和研究中心(CDER)下的OTC评价部(Office of OTC Drug Evaluation)便展开了大规模、长时间的OTC审评工程(OTC drug review)[6]。这次审评是美国有史以来对OTC安全性、有效性、标识正确性进行的一次最为全面和系统的评价。由于市场上所涉及的OTC多达35万种,如果对每一种产品进行审评,会历时过长且不具备可操作性。因此,FDA的评价战略是对这些产品中所含有的700多种活性成分进行评价,制定一个适合含有该成分的所有产品的标准,即“相对于活性成分的标准”[7]。本次审评历时十余年,FDA在对700多种活性成分按26个大的治疗类别进行划分并完成审评后,制定了针对特定类别的《OTC药品最终专论》,这些最终法规会以成文的形式收录于《联邦法典》(Code of Federal Regulations,CFR 331-358)[8],从而最终完成OTC专论制度(Monograph system)的构建。美国分类别OTC专论详见表1。
1.3 美国OTC专论的作用
1.3.1 简化审评程序,加速药品上市 美国OTC专论的全部内容被收录于联邦法规中(CFR 331-358),因此,就其法律地位而言,专论是具有约束力的实质性条款(Binding substantive rule),可以给任何满足特定药物类别下安全、有效、无标注错误的OTC提供合法上市的资格,这也是除新药申请外,OTC可以采取的另一条上市途径。在这条路径下,无论是新上市的OTC或是由处方药转化而来的OTC,只要其符合既定适应证下OTC专论的全部要求,就不用再进行新药申请,只需要向FDA备案并取得药品登记号,就可以上市销售[9]。
1.3.2 适度放宽标准,促进产品创新 美国OTC专论并未写明各类药品的最终处方,而是仅对药品的活性成分作出规范,非活性成分部分僅需证明这些成分是安全的且不影响最终产品的有效性检验即可[10]。OTC专论在一定程度上放宽了OTC上市口径,使得生产企业不仅可以快速上市简单仿制类产品,还可以根据市场情况在口味、剂型等方面进行部分改良创新,从而更好地满足了消费者多样化的产品需求,也有利于降低行业的监管负担。
2 美国OTC专论的特点分析
美国OTC专论初期构建将安全性和有效性视为唯一标准,对安全有效的追求可体现在形成过程、专论内容、增补方式等多个方面,这为专论的形成奠定了良好基础,并保障了上市产品质量。随着外部环境的发展变化,OTC专论在应对挑战的同时也积极寻求变革,高效性和灵活性成为当前OTC专论改革的重要目标。
2.1 形成过程:渐进式推进,广泛征求各方意见
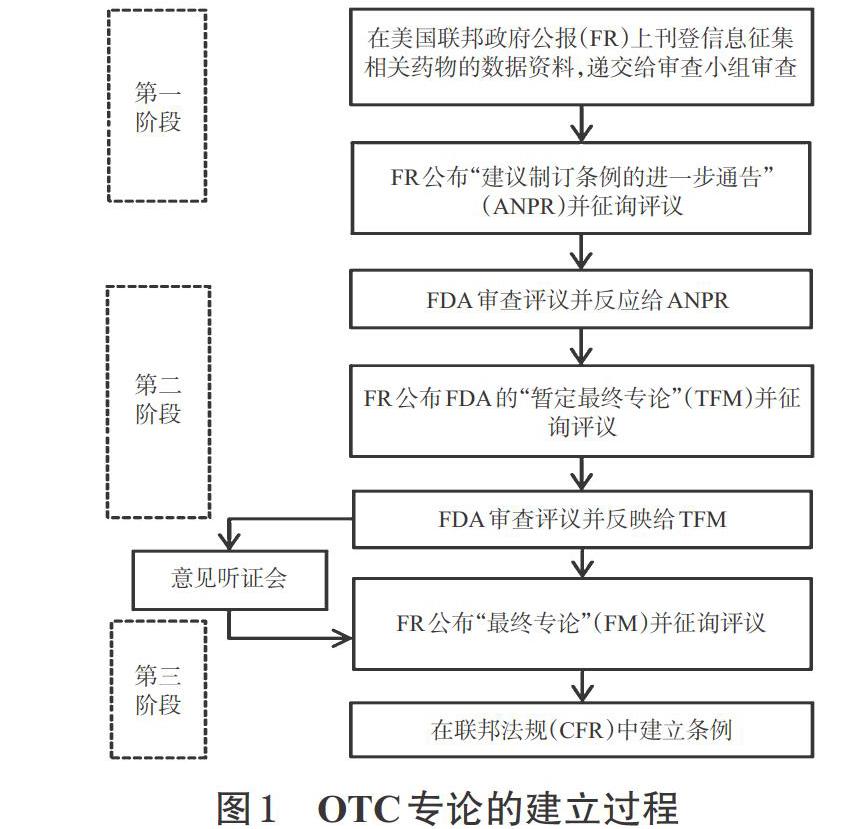
OTC专论的形成过程是一个三阶段层层推进的公共规则制定过程,是在综合监管部门专家学者、医药生产企业、广大消费者等各利益相关主体的意见上对药品安全有效标准作出科学决策的过程。OTC专论的建立过程详见图1。
整体来看,第一阶段,FDA局长在美国联邦政府公报(FR)上发布通知,收集目前已公布或未公布的药品数据资料,并组织不同治疗领域的顾问审查小组(Review panel)负责各自组内的药品评价工作。顾问审查小组会在详细分析已有资料基础上,以药品活性成分及标签内容作为研究重点,对其是否符合消费者自我药疗的安全有效标准作出评判,并将结果上报给局长审核。审核通过后,局长将以建议制订条例的进一步通告(ANPR)的形式在FR上公布顾问审查小组的评价结果,并向公众征求意见。第二阶段,FDA基于小组结论、公众意见以及可能获得的新数据,再次对每类药物中的活性成分进行评价。本次结果FDA局长将以暂定最终专论(TFM)的形式在FR上公布,并给予有关各方一段时间来提交评论或数据以回应机构提案,必要时召开听证会就争议问题进行讨论。第三阶段,局长对所有意见,包括所有新的资料和信息在内的全部内容进行审查和考虑后,在FR上公布最终专论(FM)[11]。FM中确立了某类OTC或特定OTC产品被认为是安全、有效、无标注错误的条件,可以作为药品上市的重要依据。这种渐进式审评方式最大程度上实现了药品安全有效保障的需求,为后期OTC的快捷审评奠定了良好基础。
2.2 专论内容:要点突出,细化规范指导要求
美国OTC专论明确了在美国上市OTC可被认为安全、有效、标记正确的标准[12]。OTC专论内容一般由4个部分组成:(1)总则条款(General provisions);(2)活性成分(Active ingredients);(3)标签(Labeling);(4)检验方法(Testing procedures)。其中活性成分和标签信息是OTC专论的要点,也是那些旨在通过OTC专论方式上市的OTC企业在生产过程中应当特别关注的重要内容。美国OTC专论内容详见表2。
就活性成分来看,OTC专论不仅列出了在规定浓度下,或在规定浓度范围内可以使用的药品活性成分/活性成分组合的化学名称,还对其在该治疗领域下的浓度和剂量标准作出了明确规定。当满足浓度和剂量要求时,一个产品可由该部分所列出的任何一种活性成分或活性成分组合组成;关于标签部分,则从药品身份说明(Statement of identity)、适应证信息(Indications)、使用药品前应当声明的警告(Warnings)、有关服用间隔时间和剂量要求等使用说明(Directions),以及部分供专业人员使用的标签(Professional labeling)等方面进行规范[13]。只有产品按照OTC专论的标签要求进行标示和说明,才能在满足FDA对标注无错误规定的条件下顺利上市。
2.3 增补方式:设立机制,严格规范增补条件
设立增补机制是为完善OTC专论体系而采取的进一步措施。对于没有列入OTC专论的活性成分、新适应证或新剂型,可以通过提交公民请愿书或以历时及覆盖范围申请(Time and extent applications,TEA)的方式向FDA提出申请,但是需要提交充足的安全有效证明作为依据。以TEA方式为例,其提供了一个将国外已具有多年销售和使用经验的OTC纳入美国专论的途径,根据要求,申请人需要提交有关药物的基本信息、将该药物作为OTC销售的国家名单及相关信息、该药物在这些国家的上市年限及销售信息、将该药物作为处方药销售的国家名单及相关信息、将该药物从市场上取缔或取消其OTC身份的国家名单及相关信息等,这些资料用于帮助判定所申报药品是否具备OTC资格。评价通过后,申请人仍需提交该药品的安全性和有效性数据,监管部门会在公开征集资料的基础上评估该药品是否可以被公认为安全、有效、无标注错误,并进一步决定是否修订当前OTC专论[4]。
2.4 应对挑战:简化流程,增加审评资源投入
OTC专论制定过程是一项规模庞大、复杂的系统性工程,在当前产品创新不断发展的背景下,专论制度却不断面临新的挑战。受到审评工作庞大、审评过程复杂、审评资源有限的影响,专论无法实现及时修订以应对不断变化的技术要求和新出现的安全问题,或适应产品创新或市场变化的要求。初期为确保安全性和有效性制定的药品审评制度已趋于陈旧,导致OTC专论规则制定时间过长;而在面对紧急安全问题时,仅能通过漫长的立法方式对OTC专论进行修订,缺乏时效性和灵活性。
对此FDA积极寻求变革,一方面着手简化审评流程,另一方面和行业共同制定了“OTC专论使用者付费计划”,旨在解决FDA有限的资源问题。基于该付费计划,OTC制造商在提交使用OTC专论的命令请求后,需要支付额外的费用以资助FDA进行药物审评活动。这些资源投入将主要用于OTC专论审评绩效评估、员工招聘培训、信息技术平台开发等活动,从而在保证药品安全有效的前提下缩短审评时间,完善专论规则,提升专论制度的高效性和靈活性,以便更好应对技术创新和安全问题[14]。
3 基于美国OTC专论对我国OTC注册的思考
3.1 OTC专论对于OTC注册审评的必要性
一般OTC上市有两种途径:一是通过新药申请,二是以专论方式上市,这两种方式互为补充。从功能性角度看,新药申请主要用于审批含新活性成分、新剂型、新复方等具有一定安全性风险的创新药物,而OTC专论主要适合以仿制或辅料变更为主的安全性风险可控的产品。OTC分类审批使生产企业可以根据自身情况合理选择上市渠道,从而发挥分流作用,缩短审评时间。从资源利用角度看,OTC专论制度无需经过复杂的注册审批流程即可帮助大量符合要求的仿制药产品快速上市,而监管机构也可将更多审评资源集中到新药申请过程,优化审评决策,实现资源的高效利用。从OTC特点来看,审核某个药品是否可以作为OTC使用,是审核已有的资料和经验,而不是重新进行临床试验或研究[3],而OTC专论正是对某些OTC已经通过广泛使用得到充分验证的安全性和有效性的系统总结,因此应当被纳入OTC注册体系。
当前我国OTC的技术标准和审批流程与处方药一致。对仿制类或简单创新的OTC而言,过于严苛的技术要求和繁琐缓慢的审批程序不仅阻碍了药品快速上市推广,也增加了重复性审批工作,造成了资源浪费,更不符合国际社会对OTC管理的一般认知[15],因此有必要借鉴国外经验对OTC专论进行深入研究。
3.2 当前我国实施OTC专论所面临的障碍
尽管理论上应当将OTC专论作为OTC注册审评的一种方式,但在具体的构建和实施过程中仍需结合本国国情谨慎对待。参比美国,其OTC专论的制定是伴随着FDA对1962年以前上市药品有效性再评价工作而展开的,完成了对当时已上市的所有OTC的大规模质量审评,可视为对美国OTC市场的一次全面“清洗”,具有彻底性和全面性。其产生既符合当时监管需要,又受惠于FDA丰富的审评资源,同时还有赖于监管技术的保障。
相较之下,我国目前虽然存在对OTC专论的市场需求,也有人大代表、行业企业及行业协会提出借鉴国外经验引入专论制度的重要性,但尚缺乏监管层面指导。同时受限于有限的药品审评资源,我国不能采取美国大规模激进式的审评方式,否则长期投入方式将撕裂资源的整体协调性,为监管带来更多挑战。此外,OTC专论制度可视为药品注册风险后移的一种管理方式,美国对符合专论的产品采取备案制且给予OTC制造商小部分创新权利,这需要通过后期不良反应监测来强化对前端的控制,但目前我国仿制药市场鱼龙混杂,药品风险管理体系仍有待进一步完善,难以保证前端放权之后的产品质量问题,尚不具备建设和实施OTC专论的条件。只有在优化我国资源配置和基础设施、结合本国OTC管理经验和特点的基础上,OTC专论制度才能在我国有效实施。
4 对于我国实施OTC专论的几点建议
猜你喜欢
电影文学(2016年16期)2016-10-22
中国市场(2016年36期)2016-10-19
商场现代化(2016年22期)2016-10-18
商(2016年27期)2016-10-17
电影文学(2016年9期)2016-05-17
