
猪苓多糖的PMP柱前衍生化-HPLC指纹图谱研究
2020-04-20 11:09赖戈娜贾文玉罗思婉周昌园黎雄张娴曾星
中国药房 2020年7期
赖戈娜 贾文玉 罗思婉 周昌园 黎雄 张娴 曾星
摘 要 目的:建立豬苓多糖的柱前衍生化-高效液相色谱(HPLC)指纹图谱,并测定其主要单糖组分含量,为猪苓药材的质量评价提供参考。方法:11批不同产地猪苓药材经水提醇沉、Sevage除蛋白后得到猪苓多糖。多糖经三氟乙酸(TFA)水解和1-苯基-3-甲基-5-吡唑啉酮(PMP)衍生化后,进行HPLC分析。色谱柱为HypersiL BDS C18,流动相为0.1 mol/L磷酸盐缓冲液(pH 6.84)-乙腈(84 ∶ 16,V/V),检测波长为254 nm,柱温为30 ℃,流速为1 mL/min,进样量为20 ?L。采用《中药色谱指纹图谱相似度评价系统》(2012A版)建立11批多糖样品的指纹图谱并进行相似度评价,通过与对照品比对进行色谱峰指认,再采用SPSS 23.0软件进行聚类分析,并测定多糖中主要单糖组分的含量。结果:11批猪苓多糖样品HPLC指纹图谱中共呈现出3个共有峰,分别指认为甘露糖、葡萄糖和半乳糖,各批次样品的相似度均大于0.94。聚类分析将11批多糖样品分为3类,药材编号为S1~S6、S8的样品聚为一类,药材编号为S7、S10、S11的样品聚为一类,药材编号为S9的样品单独聚为一类。含量测定结果显示,11批样品中甘露糖含量为1.571~8.771 mg/g、葡萄糖含量为26.072~132.194 mg/g、半乳糖含量为3.420~36.593 mg/g。结论:本研究建立的柱前衍生化-HPLC指纹图谱方法可为猪苓的药材质量评价提供参考;不同批次猪苓多糖中单糖组成相同,指纹图谱特征峰与药材的产地无明显相关性,药材之间的单糖含量存在明显差异。
关键词 猪苓多糖;单糖;1-苯基-3-甲基-5-吡唑啉酮;柱前衍生化;高效液相色谱法;指纹图谱
ABSTRACT OBJECTIVE: To establish pre-column derivatization-HPLC fingerprint of Polyporus polysaccharide, and to determine the contents of main monosaccharide components, so as to provide reference for quality evaluation of Polyporus umbellatus. METHODS: Polyporus polysaccharide was extracted with boiling water and precipitated by ethanol and deproteinized by Sevage from 11 batches of P. umbellatus from different producing areas. The samples were firstly hydrolyzed with trifluoro-acetic acid (TFA) and then derivatized by 1-phenyl-3-methyl-5-pyrazolone (PMP). HPLC analysis was then conducted. The determination was carried out on HypersiL BDS C18 column with mobile phase composed of 0.1 mol/L phosphate buffer (pH 6.84)-acetonitrile (84 ∶ 16, V/V) by gradient elution at the flow rate of 1.0 mL/min. The detection wavelength was set at 254 nm, and column temperature was 30 ℃. The sample size was 20 ?L. The similarity of 11 batches of Polyporus polysaccharide was evaluated by using TCM Chromatographic Fingerprint Similarity Evaluation System (2012A edition), and the contents of main monosassharide components were detected. The peak was identified by comparing with the reference substance, and cluster analysis was performed by using SPSS 23.0 software. RESULTS: In HPLC fingerprints of the 11 batches of samples, 3 common peaks were identified, namely mannose, glucose and galactose. The similarity of all samples was above 0.94. Cluster analysis classified 11 batches of samples into three categories. S1-S6, and S8 were grouped into category 1; S7, S10 and S11 were grouped into category 2; S9 was individually grouped into one category. Results of content determination showed that the contents of mannose ranged from 1.571 to 8.771 mg/g; those of glucose ranged from 26.072 to 132.194 mg/g, and those of galactose ranged from 3.420 to 36.593 mg/g. CONCLUSIONS: Established pre-column derivatization HPLC fingerprints can provide reference for quality evaluation of P. umbellatus. The monosaccharide composition of different batches of Polyporus polysaccharide is the same; there is no significant correlation between fingerprint characteristic peak and the origin of herbs; there is significant difference in the content of monosaccharide of P. umbellatus.
KEYWORDS Polyporus polysaccharide; Monosaccharide; PMP; Pre-column derivatization; HPLC; Fingerprint
猪苓又名猪灵芝、野猪苓、猪屎苓、鸡屎苓,始载于《神农百草经》,是多孔菌科真菌猪苓[Polyporus umbellatus(Pers.)Fries]的干燥菌核[1]。其道地药材产自陕西,此外在河北、河南、云南、四川等地也有分布[2]。2015年版《中国药典》(一部)猪苓质量标准项只收录了麦角甾醇含量测定[1]。而文献研究表明,猪苓多糖才是猪苓的主要活性部位,具有调节免疫、抗肿瘤、抗炎、抑菌、抗突变、保肝、保肾等与猪苓临床应用相关的药理作用[3],且猪苓水提物中多糖含量远高于麦角甾醇[4],因此应将多糖作为中药猪苓药材质量控制和品质评价的指标。但目前从猪苓中制备获得的多糖结构不一,单糖组成各不相同[5-6],猪苓传统用药形式水煎液中多糖结构特征不明。多糖一般是由多种比例相对固定的单糖构成,通过检测中药多糖的单糖组成和摩尔比,可以反映其主要特征,增加鉴别的专属性,在一定程度上对多糖进行质量控制[7-8]。指纹图谱作为中药质量控制的有效技术[9-10],目前主要是针对中药小分子成分,较少用于多糖等生物大分子,这是由大多数糖类成分无紫外吸收、分子量大、结构复杂多样特点决定的[11]。但通过分析各中药中多糖的主要单糖组成与含量,建立单糖组成指纹图谱,可以鉴别不同来源的多糖;通过测定其主要组成单糖的绝对含量,可整体评价不同批次药材质量的优劣。因此,本研究采用1-苯基-3-甲基-5-吡唑啉酮(PMP)柱前衍生化-高效液相色谱(HPLC)法分析猪苓水煎液多糖的单糖组成和含量,并运用指纹图谱技术比较不同产地猪苓多糖中单糖组成与含量差异,为中药猪苓多糖质量标准的建立提供参考。
1 材料
1.1 仪器
1200型HPLC仪(美国Agilent公司);Milli-Q型超纯水仪系统(美国Millipore公司);Alpha2-4LSC-Plus型冷冻干燥机(德国Martin Christ公司);RV 10 basic V型旋转蒸发仪(德国IKA公司);AB135-S型十万分之一天平、ET58型pH计(德国Mettler-Toledo公司)。
1.2 药品与试剂
葡萄糖对照品(批号:S10S9I69833,纯度:≥98%)、甘露糖對照品(批号:C25D8H51117,纯度:≥98%)、半乳糖对照品(批号:Z22J9H64187,纯度:≥98%)均购自上海源叶生物科技有限公司;PMP(美国Aladdin公司,批号:C1822095);三氟乙酸(TFA,德国Sigma公司,批号:STBF6942V);乙腈为色谱纯,其余试剂均为分析纯,水为自制超纯水。
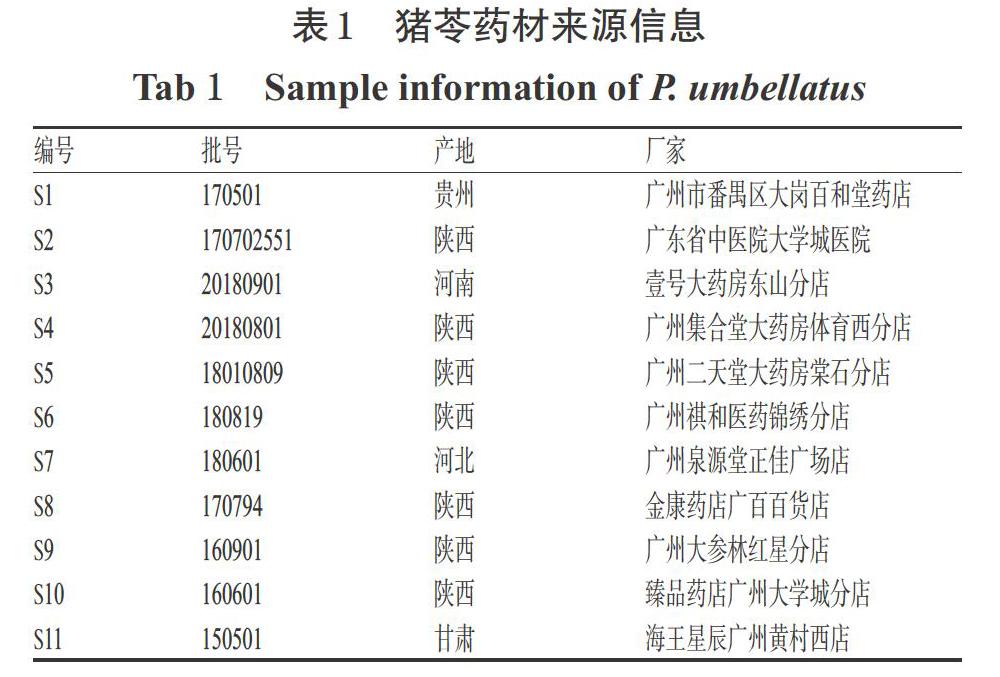
11批猪苓原药材分别购自不同厂家,经广州中医药大学第二临床医学院黎雄副研究员鉴定均为多孔菌科真菌猪苓[P. umbellatus(Pers.)Fries]的干燥菌核,样品保存于广州中医药大学第二临床医学院Ⅰ期临床研究分析室。11批猪苓药材来源信息见表1。
2 方法与结果
2.1 猪苓多糖的提取和纯化
称取各批次猪苓药材50 g,粉碎后过60目筛,以料液比为1 ∶ 10(g/mL)的比例加水回流提取2次,每次1.5 h,合并滤液,减压浓缩至200 mL;加入无水乙醇至乙醇体积分数为80%,静置过夜;以1 000 r/min离心10 min,沉淀加适量水溶解后,按4 ∶ 1(V/V)的比例加入Sevage试剂[氯仿-正丁醇(4 ∶ 1),V/V],搅拌混匀,去除下层溶剂和中间层蛋白,反复多次,直至蛋白除尽,冷冻干燥后即得猪苓多糖。采用苯酚-硫酸法[10],以无水葡萄糖为标准品测定11批次猪苓多糖纯度,得到各批次药材样品(药材编号:S1~S11)多糖纯度分别为43.00%、54.51%、56.05%、52.20%、58.08%、56.60%、60.85%、45.71%、51.19%、50.30%、57.74%。
2.2 溶液的制备
2.2.1 混合对照品溶液的制备 精密称取甘露糖对照品5.08 mg、葡萄糖对照品4.99 mg、半乳糖对照品5.07 mg,分别用水溶解后定容至5 mL量瓶中,充分摇匀,得各成分单一对照品溶液。分别取各单一对照品溶液1 mL至同一10 mL量瓶中,加水定容至刻度,即得混合对照品溶液。
2.2.2 供试品溶液的制备 精密称取猪苓多糖20 mg于安瓿瓶中,加入3 mol/L的TFA溶液6 mL溶解,于110 ℃下水解6 h,放冷后,减压浓缩至干;再加入1 mL甲醇,蒸干,重复4次以除尽TFA;干燥物加入20 mL水溶解,即得供试品溶液,保存备用。
2.2.3 阴性对照溶液的制备 称取20 mg猪苓多糖(药材编号:S1)溶于20 mL水中,制备成未经水解的阴性对照溶液。
2.3 衍生化处理
分别精密移取“2.2”项下混合对照品溶液、供试品溶液、阴性对照溶液各500 ?L,加入0.3 mol/L的NaOH溶液200 ?L、0.5 mol/L的PMP甲醇溶液200 ?L,充分混匀,在70 ℃水浴中衍生化反应60 min;放冷,加入0.3 mol/L的HCl溶液200 ?L中和NaOH,再加入氯仿1 mL充分萃取,重复萃取3次后,取上层水层液,经0.22 ?m滤膜滤过,即得各衍生化溶液。
2.4 色谱条件与系统适用性试验
色谱柱:HypersiL BDS C18(250 mm×4.6 mm,5 ?m) ;流动相:0.1 mol/L磷酸盐缓冲液(pH 6.84)-乙腈(84 ∶ 16,V/V);检测器:紫外检测器;检测波长:254 nm;柱温:30 ℃;流速:1 mL/min;进样量:20 ?L。取按“2.3”项下方法衍生化处理的混合对照品溶液、供试品溶液(药材编号:S1)和阴性对照溶液,分别按上述色谱条件进样分析,记录色谱图。结果,在该色谱条件下,各成分均能达到基线分离,各成分峰间分离度均大于1.5,理论板数以甘露糖计不低于8 000,且阴性对照溶液不干扰样品峰的测定。色谱图详见图1。
2.5 HPLC指纹图谱的建立与分析
2.5.1 精密度试验 取“2.2.1”项下混合对照品溶液,按“2.3”项下方法进行衍生化后,按“2.4”项下色谱条件连续进样6次,记录峰面积。以葡萄糖峰(峰2)为参照峰,计算得各共有峰相对保留时间的RSD均小于1%(n=6)、相对峰面积的RSD均小于3%(n=6),表明仪器精密度良好。
2.5.2 重复性试验 精密称取同一批号(药材编号:S1)的猪苓多糖样品6份,按“2.2.2”项下方法制备供试品溶液后,再按“2.3”项下方法进行衍生化,然后按“2.4”项下色谱条件进样测定,记录峰面积。以葡萄糖峰(峰2)为参照峰,计算得各共有峰相对保留时间的RSD均小于1%(n=6)、相对峰面积的RSD均小于3%(n=6),表明该方法重复性良好。
2.5.3 稳定性试验 称取猪苓多糖样品(药材编号:S1)适量,按“2.2.2”项下方法制备供试品溶液后,再按“2.3”项下方法进行衍生化,然后分别于室温条件下放置0、2、4、8、10、12 h时按“2.4”项下色谱条件进样测定,记录峰面积。以葡萄糖峰(峰2)为参照峰,计算得各共有峰相对保留时间的RSD均小于1%(n=6)、相对峰面积的RSD均小于3%(n=6),表明供试品溶液在室温下放置12 h内稳定。
2.5.4 指纹图谱的生成 取11批猪苓多糖样品,分别按“2.2.2”项下方法制备供试品溶液后,再按“2.3”项下方法进行衍生化,然后按“2.4”项下色谱条件进样测定,记录色谱图。将11批样品的色谱图导入《中药色谱指纹图谱相似度评价系统》(2012A版)中,选择S1号样品的图谱作为参照图谱(S1号图谱各峰分离完全、信号强度大),设置时间窗宽度为0.2 min,图谱间距为40,通过多点校正及自动匹配生成11批衍生化处理的猪苓多糖的叠加指纹图谱,并采用中位数法生成对照指纹图谱。结果,共得到3个共有色谱峰。11批衍生化处理的猪苓多糖的叠加指纹图谱详见图2,对照指纹图谱详见图3。
2.5.5 相似度评价 以对照指纹图谱为参照,采用《中药色谱指纹图谱相似度评价系统》(2012A版)进行相似度评价。结果,11批次猪苓多糖的单糖组成的相似度均在0.94以上,表明药材样品间差异较小,质量稳定性良好。11批样品的相似度评价结果详见表2。
2.5.6 色谱峰的指认 通过将HPLC对照指纹图谱与对照品溶液的HPLC图谱(见图1A)比对,指认了峰1为甘露糖、峰2为葡萄糖、峰3为半乳糖。因峰2的分離度良好、峰面积大且稳定,故以其保留时间和峰面积为参照,计算得各共有峰相对保留时间的RSD为0~0.29%(n=11),相对峰面积的RSD为0~39.85%(n=11)。结果表明,不同批次的猪苓多糖虽然具有相同的单糖组成,但是含量差异较大。11批样品中共有峰的相对保留时间测定结果详见表3,相对峰面积测定结果详见表4。
2.5.7 聚类分析 将11批猪苓多糖样品共有峰的相对峰面积导入SPSS 23.0软件中,以平方欧氏距离作为分类依据,采用Ward法聚类。结果显示,当欧氏距离为5时,11批猪苓多糖可聚为3类,其中药材编号为S1~S6、S8的样品聚为一类,相似度≥0.995;药材编号为S9的样品单独聚为一类,相似度在0.995~0.999之间;药材编号为S7、S10、S11的样品聚为一类,相似度在0.946~0.990之间。以上结果表明,不同来源的猪苓多糖在化学组成方面具有很好的相似性。11批猪苓总多糖的聚类分析树状图详见图4。
2.6 单糖含量的测定
2.6.1 线性关系考察 精密吸取“2.2.1”项下甘露糖、葡萄糖、半乳糖单一对照品溶液各500 ?L,置于同一5 mL量瓶中,加水定容至刻度,充分混匀。分别吸取上述混合溶液500、250、125、100、62.5、50、25、12.5 ?L至4 mL离心管中,加水稀释至500 ?L,按“2.3”项下方法进行衍生化,制备系列质量浓度的样品溶液,然后分别按“2.4”项下色谱条件进样测定,记录峰面积。以3种单糖的质量浓度(x,?g/mL)为横坐标、峰面积(y)为纵坐标进行回归分析。结果,甘露糖、葡萄糖、半乳糖在各自质量浓度范围内线性关系均良好(r均大于0.999 0),详见表5。
2.6.2 定量限和检测限 取“2.2.1”项下混合对照品溶液,加水倍比稀释,按“2.3”项下方法进行衍生化,然后按“2.4”项下色谱条件进行测定,以信噪比10 ∶ 1、3 ∶ 1分别计算定量限、检测限。结果,甘露糖、葡萄糖、半乳糖的定量限分别为0.045 6、0.092 4、0.092 2 μg/mL,检测限分别为0.022 8、0.030 8、0.026 3 μg/mL。
2.6.3 重复性试验 精密称取同一批样品(药材编号:S1)6份,按“2.2.2”项下方法制备供试品溶液,再按“2.3”项下方法进行衍生化,然后按“2.4”项下色谱条件进样测定,记录峰面积,并根据回归方程计算样品中3种单糖的含量。结果,甘露糖、葡萄糖、半乳糖的平均含量分别为3.568、47.332、17.671 mg/g,RSD分别为4.33%、2.77%、3.32%(n=6),表明本方法的重复性良好。
2.6.4 稳定性试验 取“2.3”项下同一批衍生化处理的供试品溶液(药材编号:S1),分别于室温下放置0、2、4、8、10、12 h时按“2.4”项下色谱条件进样测定,记录峰面积。结果,甘露糖、葡萄糖、半乳糖峰面积的RSD分别为4.81%、4.30%、4.49%(n=6),表明该溶液在室温条件下放置12 h内稳定性良好。
2.6.5 加样回收率试验 取“2.2.2”项下已知含量的供试品溶液(药材编号:S1)6份,每份1 mL,每份加入15.45 μg/mL的甘露糖、425.68 μg/mL的葡萄糖、95.46 μg/mL的半乳糖对照品溶液各1 mL(按“2.2.1”项下方法重新制备),按“2.3”项下方法进行衍生化后,再按“2.4”项下色谱条件进样测定,记录峰面积,根据回归方程计算各成分的含量并计算其回收率。结果,甘露糖、葡萄糖、半乳糖的平均加样回收率分别为99.44%、98.77%、97.57%,RSD分别为2.01%、1.66%、2.03%(n=6),表明该方法准确度较好,详见表6。
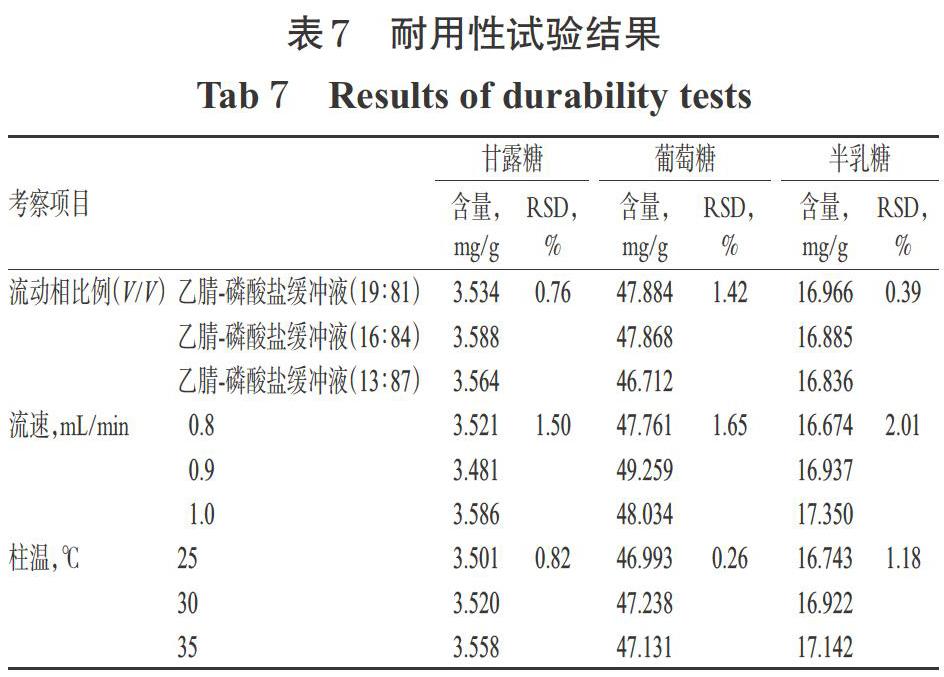
2.6.6 耐用性试验 取“2.2.2”项下供试品溶液(药材编号:S1)适量,按“2.3”项下方法衍生化,在其它色谱条件同“2.4”项下不变的情况下,考察不同流动相比例[乙腈-磷酸盐缓冲液(19 ∶ 81、16 ∶ 84、13 ∶ 87,V/V)]、不同流速(0.8、0.9、1.0 mL/min)、不同柱温(25、30、35 ℃)对3种单糖含量测定结果的影响。结果,3种变化条件下甘露糖含量的RSD分别为0.76%、1.50%、0.82%(n=3),葡萄糖含量的RSD分别为1.42%、1.65%、0.26%(n=3),半乳糖含量的RSD分别为0.39%、2.01%、1.18%(n=3),RSD均小于3%,表明本方法的耐用性较好,详见表7。
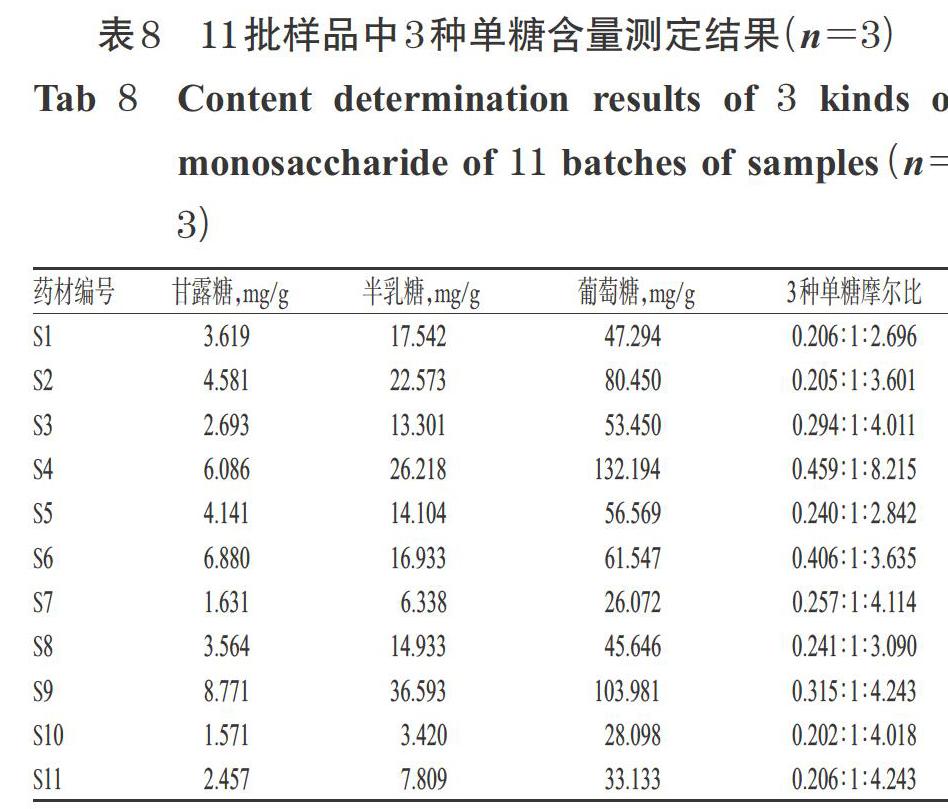
2.6.7 样品含量测定 按上述所建立的PMP柱前衍生化HPLC法测定11批次猪苓多糖各单糖含量,每个样品重复测定3次。结果,11批样品中甘露糖含量在1.571~8.771 mg/g之间、半乳糖含量在3.420~36.593之间、葡萄糖含量在26.072~132.194 mg/g之间;不同批次多糖的单糖含量存在明显差异,但葡萄糖含量均为最高,其次是半乳糖,含量最低的为甘露糖。以半乳糖的摩尔质量为基准1计算各批次多糖的单糖组成摩尔比,得到甘露糖、半乳糖、葡萄糖的摩尔比在(0.202~0.459) ∶ 1 ∶ (2.696~8.215)之间。11批样品中3种单糖含量测定结果详见表8。
3 讨论
本课题组前期选择TFA和PMP的浓度分别为3、0.5 mol/L,以单糖的峰面积为评价指标,对酸水解的时间以及PMP衍生化的时间和温度进行了筛选,得到了最佳衍生化条件,即酸水解6 h、70 ℃水浴下衍生化60 min。此外,本课题组前期比较了各单糖在HypersiL BDS C18(250 mm×4.6 mm,5 ?m)、Agilent ZORBAX Eclipse XDB-C18(150 mm×4.6 mm,5 ?m)两种不同类型色谱柱上的分离效果,结果发现,长柱比短柱的分离效果好,短柱上葡萄糖峰和半乳糖峰不能完全分离。同时,本课题组前期还考察了0.02、0.05、0.1 mol/L磷酸盐缓冲液对样品分离效果的影响,结果使用0.02、0.05 mol/L的磷酸盐缓冲液时色谱峰不对称、拖尾严重;而在0.1 mol/L磷酸盐缓冲液下样品色谱峰峰形对称、分离度好。
本研究以不同产地的11批猪苓药材为样品,采用传统水煎工艺制备总多糖,Sevage法除蛋白后采用PMP柱前衍生化-HPLC法測得其主要单糖组成均为甘露糖、葡萄糖和半乳糖,所测得的猪苓多糖的单糖成分种类较少。分析其原因,可能是未纯化的粗多糖中本身含有部分游离单糖,采用Sevage法去除粗多糖中蛋白的同时,也可能除去了游离的单糖和含蛋白比例高的糖肽。本研究基于PMP柱前衍生化-HPLC法建立了11批猪苓多糖的指纹图谱,采用《中药色谱指纹图谱相似度评价系统》(2012A版)计算这11批样品的相似度后发现,样品相似度均在0.94以上,这说明不同来源的猪苓多糖样品在化学组成方面具有良好的相似性,该制备方法得到的猪苓多糖的质量较为稳定。聚类分析将11批猪苓多糖分为3类,分类结果与产地无相关性,说明产地因素对多糖的单糖组成影响较小,但这一结论仍需更多产地、批次的样品进一步验证。通过测定猪苓多糖的3种主要单糖含量发现,不同批次猪苓多糖样品中各单糖含量及单糖摩尔比存在较大差异,但各批次猪苓多糖中单糖含量最高的均为葡萄糖,其次是半乳糖和甘露糖,而单糖的含量差异是否影响其药理活性,还有待进一步研究。
参考文献
[ 1 ] 国家药典委员会.中华人民共和国药典:一部[S]. 2015年版.北京:中国医药科技出版社,2015:318-319.
[ 2 ] 任洁,李太元,李艳茹,等.长白山猪苓与陕西猪苓的菌丝体转录组差异比较分析[J].延边大学农学学报,2019,41(3):1-8.
[ 3 ] 夏琴,李敏,周进,等.不同产地、商品规格及生长年限猪苓麦角甾醇及多糖的含量分析[J].中药材,2015,38(1):45-48.
[ 4 ] 王天媛,张飞飞,任跃英,等.猪苓化学成分及药理作用研究进展[J].上海中医药杂志,2017,51(4):109-112.
[ 5 ] HE P,ZHANG A,ZHANG F,et al. Structure and bioactivity of a polysaccharide containing uronicacid from Polyporus umbellatus sclerotia[J]. Carbohydr Polym,2016.DOI:10.1016/j.carbpol.2016.07.010.
[ 6 ] HE PF,HE L,ZHANG AQ,et al. Structure and chain conformation of an eutral polysaccharide from sclerotia of Polyporus umbellatus[J]. Carbohydr Polym,2017. DOI:10.1016/j.carbpol.2016.08.041.
[ 7 ] 李运,邱国玉,李晓春,等.基于主成分分析的前胡类药材HPLC指纹图谱研究[J].药物分析杂志,2019,39(7):1323-1329.
[ 8 ] 杜泽飞,陶爱恩,夏从龙,等.基于PMP-HPLC和化学计量学的黄精基原物种多糖差异分析[J].中国实验方剂学杂志,2019,25(15):25-29.
[ 9 ] 邹纯才,鄢海燕.我国中药色谱指纹图谱相似度评价方法30年(1988-2017年)研究进展与展望[J].中国中药杂志,2018,43(10):1969-1977.
[10] 汪海斌,石岩,李芳,等.中药黄芪指纹图谱的研究进展[J].中国药房,2017,28(33):4749-4752.
[11] 李贝贝,卢燕.多糖色谱指纹图谱的研究方法进展[J].药学研究,2018,37(1):41-45.
(收稿日期:2019-11-18 修回日期:2020-01-15)
(编辑:林 静)
猜你喜欢
国际传播(2022年1期)2022-05-10
ELLE世界时装之苑(2021年10期)2021-10-11
科学与财富(2021年34期)2021-05-10
科学与财富(2021年36期)2021-05-10
分析化学(2018年4期)2018-11-02
新城乡(2018年6期)2018-07-09
科技视界(2018年1期)2018-03-15
分析化学(2017年12期)2017-12-25
湖北农业科学(2017年1期)2017-03-09
科学24小时(2016年10期)2016-10-20
