
Hydrogen-Bond Induced Crystallization of Silicalite-1 Zeolite as Revealed by Solid-State NMR Spectroscopy
2020-04-24 05:42XiaolongLiuQiangWangChaoWangJunXuFengDengStateKeyLaboratoryofMagneticResonanceandAtomicandMolecularPhysicsNationalCentreforMagneticResonanceinWuhanWuhanInstituteofPhysicsandMathematicsInnovationAcademyforPrecisionMeas
物理化学学报 2020年4期
Xiaolong Liu , Qiang Wang , Chao Wang , Jun Xu ,*, Feng Deng ,* State Key Laboratory of Magnetic Resonance and Atomic and Molecular Physics, National Centre for Magnetic Resonance in Wuhan, Wuhan Institute of Physics and Mathematics, Innovation Academy for Precision Measurement Science and Technology,Chinese Academy of Sciences, Wuhan 43007, P. R. China.
2 School of Materials, Sun Yat-Sen University, Guangzhou 510275, P. R. China.
Abstract:The flexible chemical composition of the frameworks with tunable pore size and geometry of molecular dimensions makes zeolites widely used in chemical and petrochemical industry fields. The understanding of crystallization mechanism is important for a rational design of new zeolite with target structure and property, which however is still a big challenge in the field of material science. In this work, the specific spatial correlations/interactions between the SiO−··HO ― Si hydrogen bonds within the charged framework of silicalite-1 (MFI topology)zeolite and the alkyl chains of tetrapropylammonium ion (TPA+) organic structure direction agents (OSDAs) were studied by one-dimensional (1D) and two-dimensional (2D) solid state-NMR spectroscopy in combination with other techniques,with the aim to shed light into the crystallization mechanism of silicalite-1. The “solvent-free” route was used to study the crystallization process. Silicalite-1 crystals were also prepared following the hydrothermal synthesis route. The structural properties of as-synthesized TPA-silicalite-1 samples during the crystallization were characterized by XRD and scanning electron microscopy (SEM) images, which showed the evolution of long-range periodic structure and cyrtal growth. The 1H-29Si CP/MAS NMR experiments showed that the reorganization of the silica or silicates occurred in the crystallization process. The lH-13C CP/MAS NMR experiments performed on the samples synthesized with different time indicated that the TPA+ ions in the amorphous samples experienced a constrained environment, forming the inorganic-organic composites. The splitting of the methyl carbon signal from TPA+ ions was observed in the 13C NMR spectra, which is the direct reflection of the interactions between the methyl groups and the silicate framework in the straight and zig-zag channels of silicalite-1. Two types of SiO−··H―OSi hydrogen bonds (SiO−··H―OSi hydrogen bond in-cage and SiO−··H―OSi hydrogen bond between lamellae) have been identified by 2D 1H double quantum (DQ)-single quantum (SQ) MAS NMR and 2H MAS NMR during the crystallization of silicalite-1. The SiO−··H―OSi hydrogen bonds between lamellae are formed and gradually transformed into the in-cage ones during the crystallization process. Their functions have been revealed in the formation of silicalite-1: the SiO−··H ― OSi hydrogen bond in-cage provides the stereoscopic counterbalance for the positive charges from TPA+ ions and this stereoscopic electrostatic interaction is the key factor to transform inorganic-organic composites with the MFI structure property, even though the long-range periodic MFI structures have not been established yet; the SiO−··H―OSi hydrogen bond between lamellae acts as a connector to assemble the silicate species together to generate the zeolite framework. 2H MAS NMR spectra show that the SiOH nests exist in the zeolite framework even though the long-range periodic structures have been fully established.
Key Words:Zeolite;Crystallization mechanism;Solid-state NMR;Structure direction agent;Hydrogen bond
1 Introduction
Zeolites are microporous crystalline inorganic materials with well-defined pore systems. The flexible chemical composition of the frameworks with tunable pore size and geometry of molecular dimensions makes zeolites widely used in chemical and petrochemical industry fields as catalysts, adsorbents and separators1,2. The synthesis process of ZSM-5 (MFI topology)has been extensively studied due to its academic and commercial importance.
The tetrapropylammonium (TPA+) cations are usually used as the organic structure direction agents (OSDAs) to synthesize ZSM-5 and are located at the channel intersections formed by parallel straight channels and zig-zag channels in the crystal structures of ZSM-5, with their propyl chains extending into both the straight and zig-zag channels3,4. The structural directing roles of TPA+cations involve preorganization of silicate species around the TPA+cations to yield the inorganic-organic composites and then assembly of these inorganic-organic composites into ZSM-5 crystals with occluded TPA+cations in the channel intersections5–7. By using1H-29Si cross polarization magic angle spinning (CP/MAS) solid-state NMR technique,Davis and co-workers confirmed that short-range intermolecular interactions (on the order of van der Waals contact distances) are established by the formation of inorganic-organic composites during heating of the zeolite synthesis gel prior to the full development of long-range ordered ZSM-5 crystalline structure8,9.However, in the crystallized ZSM-5, the positive charges of TPA+cations are always balanced by the negative charges from F−anions or the SiO−··H―OSi hydrogen bonds in silicalite-110–12.Using two-dimensional (2D)2H–{1H} NMR correlation NMR spectroscopy, Shantz and Lobo observed that the electrostatic organic-inorganic interactions influence the location of SiO−··H―OSi hydrogen bonds in the zeolite framework13,14. In addition, through studying the spatial correlations between the T3+(i.e., Al3+) atoms and the organic structure directing agents,Lobo and co-workers proposed that during the zeolite synthesis process the charge distributions of OSDAs control the location of T3+atoms in the zeolite framework13,14. Therefore, it is reasonable to expect that during the assembly-to-crystallization stage the electrostatic interactions between the OSDAs and the SiO−··H―OSi hydrogen bonds or F−within the framework play more important structural direction role in the formation of the periodical long range ZSM-5 crystals with respect to the van der Waals interactions.
The crystallization mechanism of industrially important zeolites, such as ZSM-5 and Beta, is of great scientific significance because it can provide structural information that is critical to fully understanding their physicochemical properties and catalytic functions, which should also be very helpful for improving the performance of these materials. In the last five years, some wonderful studies on the crystallization processes of zeolites have been published. For example, Raman spectroscopy has been used to study the development of TEA+conformers in zeolite Beta and ZSM-515,16; and 2D1H DQ-SQ MAS NMR techniques have been used to study the spatial correlations between the defect sites and TPA+cations17,18. Although they all provide important information on the structure direction of OSDAs in the crystallization processes of zeolites ZSM-5 and Beta, how the OSDAs direct the formation of high silica zeolite ZSM-5 is still unclear. High silica zeolites exhibit a close geometric relationship between the pore architecture and occluded organic cations, therefore the spatial correlations/interations between the charged pore walls and the occluded organic molecules are the keys to understanding the role of OSDAs.
In this work, the specific spatial correlations/interactions between the SiO−··H―OSi hydrogen bonds within the charged framework of silicalite-1 (Al-free anagous of ZSM-5) and the alkyl chains of TPA+OSDAs were studied by 2D1H doublequantum and single-quantum magic angle spinning (DQ-SQ MAS) NMR techniques, with the aim to shed light into the crystallization mechanism of silicalite-1. Two types of SiO−··H―OSi hydrogen bonds were identified and their functions in the synthesis process were revealed. Instead of hydrothermal synthesis, a new developed “solvent-free”synthesis route19was chosen to study the crystallization process of silicalite-1. Although water is not directly added to the synthesis mixtures in the “solvent-free” method, traces of water are still needed in the initial raw materials. There are two benefits with this synthesis method: one is the shorter crystallization time and the solid-state phase at the beginning of crystallization process, which makes solid-state NMR studies of the zeolite crystallization process easier than those under the hydrothermal synthesis conditions that need a much longer crystallization time and has a liquid-state starting phase for the preparation of silicalite-1; the other is that2H isotopes as the tracer of the ractant can easily be introduced into the synthesis system by mixing the anhydrous Na2SiO3with the proper amount of D2O,and thus2H MAS NMR can also be used to monitor the zeolite crystallization process. The2H MAS NMR spectra with quadrupolar coupling constant analysis provided insights into the formation and evolution of the SiO−··H―OSi hydrogen bonds during the crystallizaiton of silicalite-1. For comparison,silicalite-1 crystals were also prepared under hydrothermal synthesis condition.
2 Experimental
2.1 Crystallization of silicalite-1
The Na2SiO3·5D2O was prepared through re-crystallization of anhydrous Na2SiO3(≥ 98%) with a proper amount of D2O. In a typical synthesis, 0.67 g of Na2SiO3·5D2O, 0.16 g of fumed silica(≥ 99.8%), 0.12 g of TPABr (≥ 99.0%), and 0.22 g of NH4Cl (≥99.5%) were mixed and grinded. Then the mixture was transferred into a glass tube and sealed by flame. The sealed tubes were heated at 180oC in oven for different time intervals and then removed from the oven and cooled down to room temperature. Under the solvent-free synthesis condition, the crystallization time is quite short (ca. 24 h) and the solid raw materials are mechanically mixed and heated at 180 °C. In this synthesis route, the key factor is the utilization of NH4Cl because its dissociation products are HCl and NH3at high temperatures:NH3molecules react with H2O to produce OH−anions, and the dissociated HCl reacts with Na2SiO3to form H2SiO3(SiO2·H2O)and NaCl. The mineralizing agent (OH−) in zeolite synthesis converts the reactants into mobile and reactive silicate species,thus enabling the synthesis to proceed, and it facilitates the transformation process from totally amorphous material to ordered zeolite crystalline. During the whole temperature heating period, the solid raw materials did not melt.
Silicalite-1 crystals were also prepared following the hydrothermal synthesis route using tetraethyl orthosilicate(TEOS, Aldrich, ≥ 99.0%), alkali-free tetrapropylammonium hydroxide (TPAOH) solutions (1 mol∙L−1in water) and water.The gel composition is SiO2:0.4TPAOH:40H2O, then the final solution was transferred into a Teflon-lined stainless-steel autoclave and heated at 175 °C for 14 days under static conditions. After crystallization, the autoclave was rapidly cooled and the zeolite was recovered by centrifugation, washed with distilled water and dried at 60 °C.
2.2 Characterization
29Si and13C CP/MAS NMR experiments were performed on a Bruker AVANCE-III 500 spectrometer using a 4 mm MAS probe. All the CP MAS experiments were carried out at a spin rate of 10 kHz, using a1H π/2 pulse length of 3.85 μs, a TPPM1H decoupling of 65 kHz and a recycle delay of 5 s. The Hartmann-Hahn condition was achieved using kaolinite or hexamethylbenzene (HMB), and the contact time was set to 4 ms and 2 ms for29Si and13C CP MAS experiments respectively.29Si chemical shifts were referenced with kaolinite as an external secondary standard, by setting its resonance to −91.5 ppm.13C chemical shifts were referenced with HMB as an external secondary standard, by setting the resonance of its methyl groups to 17.35 ppm.
1H MAS and 2D1H DQ-SQ MAS NMR spectra were carried out in a 1.9 mm MAS probe on a Bruker AVANCE-III 500 spectrometer with a sample spinning rate of 40 kHz, a1H π/2 pulse length of 1.75 µs and a recycle delay of 2 s. R1225symmetry pulse was used for DQ excitation (texc)/reconversion(trec), which has been demonstrated to be an efficient homonuclear DQ recoupling scheme at fast MAS rates20. R1225symmetry pulse sequence was applied with texc= trec= 200 µs =8TR, 128 t1increments with 16 transients each were acquired with a recycling delay of 2 s. The spectra were referenced with respect to tetramethylsilane (TMS) using solid tetrakis(trimethylsilyl)silane as a secondary standard (0.27 ppm for1H). All solid samples for the DQ-SQ MAS NMR experiments had been washed with D2O and dried at 110 °C for 16 h.
2H MAS NMR experiments were performed on a Varian Infinity-plus 400 spectrometer. Quadrupolar echo NMR method was used to acquire2H MAS NMR spectra, in which the recycle delay is 1 s, the scan number is 400000 for each spectrum and the spinning rate is 8 kHz.2H chemical shifts were referenced with D2O as an external secondary standard, by setting its resonance to 4.7 ppm. Reconstruction of the spinning sidebands of the2H spectra was performed with DMFIT software21.
Powder X-ray Diffraction data was collected in transmission geometry with a Bruker D8X diffractometer using Cu Kαradiation. Diffractograms were collected between 4° and 30°(2θ) with steps of 0.02° and 1 s per step. Scanning electron microscopy (SEM) experiments were performed on Hitachi SU-1510 electron microscopes.
3 Results and discussion
The structural properties of as-synthesized samples with increasing heating times were characterized by XRD as shown in Fig. 1 (XRD patterns in the whole time range were shown in Fig. S1, Supporting Information (SI)). The scanning electron microscopy (SEM) images of these samples are given in Fig. S2(SI). The XRD peaks at 2θ = 13°, 22.4° and 25.5° are observable throughout the crystallization process, and these peaks in Fig. 1 and Fig. S1 are mainly from the raw material Na2SiO3. Because solid state sodium silicates were trapped in the crytals of silicalite-1 or fused together with silicalite-1 crystals, the XRD peaks from sodium silicates are still observed in the final products.
Fig. 1 shows powder XRD patterns of the samples collected after heat treatment for different periods of time. Upon heating up to 16 h, the appearance of XRD peaks at 2θ = 23.3°, 23.8°,24.0° and 24.5° are probably due to the formation of smaller ring structures of aluminosilicates5in the introduction and nucleation period. A notable change was observed after 16 h heating, when the XRD peaks at 2θ = 7.9° and 8.6° appeared, indicating the long-range periodic structure of silicalite-1 was established.Further increase of these peak intensities with prolonged heating treatment indicates the crystal growth of zeolite silicalite-1.
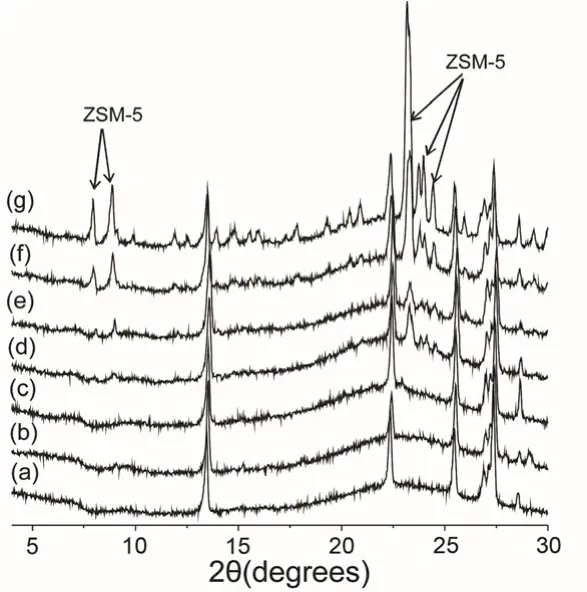
Fig. 1 XRD patterns of as-synthesized TPA-silicalite-1 samples heated at 180 °C for different time: (a) 0 h, (b) 5 h, (c) 15 h, (d) 16 h,(e) 18 h, (f) 20 h and (g) 24 h.
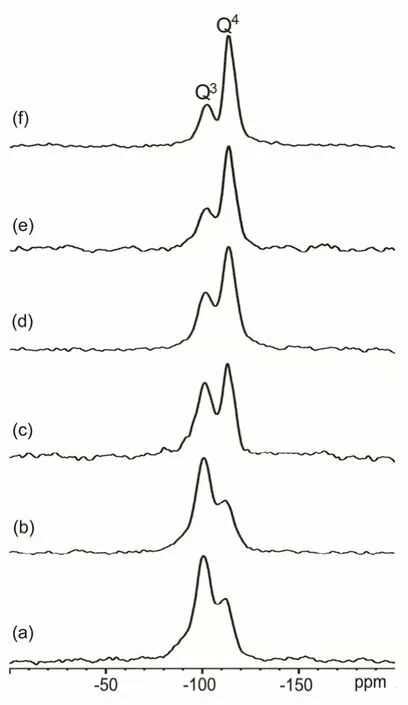
Fig. 2 1H-29Si CP/MAS NMR spectra of as-synthesized TPA-silicalite-1 samples heated at 180 °C for different time: (a) 0 h,(b) 5 h, (c) 15 h, (d) 16 h, (e) 18 h and (f) 20 h.
In order to trace the evolution of the zeolite framework,1H-29Si CP/MAS NMR experiments on the as-synthesized samples were performed. As shown in Fig. 2, two signals at −101 and−113 ppm are observable on all the samples, corresponding to Q3and Q4species (Qnrepresents Si(OSi)n(OH)4−n). At the induction period (0–15 h), the Q3/Q4signal intensity ratio decreases very rapidly compared to the crystallization rate along with XRD patterns, suggesting that the reorganization of the silica or silicates has occurred in the crystallization process during which inorganic-organic composites would be formed and serve as the basic building units for the nucleation and crystal growth of the self-assembled silicalite-1. This is evidenced by the following 2D1H DQ-SQ and2H NMR experiments. The organization of silicates by the organic molecules should require close contact between these species,which was revealed by efficient polarization transfer from1H to29Si in CP/MAS NMR spectra6. However, the1H-29Si CP/MAS NMR spectra do not show significant change of the Q3/Q4signal intensity ratio from from 16 to 20 h. Thus more structural information on the transformation during the crystal growth can not be obtained. If the self-assembly process of silicalite-1 is oriented by the organic TPA+molecules through the noncovalent interactions, the change of the environment around the TPA+ions would shed some light on the formation of silicalite-1 zeolite. ThelH-13C CP/MAS NMR experiments were performed on the samples synthesized with differnt time. As shown in Fig. 3, the13C signals show a gradual change along with the heating time. Besides the methyl signal (12.8 ppm) from the free TPA+ions, the prounced signals at 63.4 and 16.8 ppm are due to the methylene carbon adjacent to the nitrogen group and second methylene carbon, respectively. Note that the XRD pattern (Fig. 1d) shows that no long-range silicalite-1 crystals are apparently formed at this statge. This observation suggests that the TPA+ions in these amorphous samples experience a constrained environment. It is most likely that the TPA+cations were diffused into the silica species and formed the inorganicorganic composites. After 15 h heating, Fig. 3d–f clearly show the splitting signals (11.6 and 10.6 ppm) from the methyl groups in TPA+, while the signals at 63.4 and 16.8 ppm remains unchanged except for their intensities. The splitting signals may correspond to the methyl groups of TPA+occluded into the different channels, implying that the channel systems have been fully established. The spectral signature of occluded TPA+reflects the changes in conformation and the intermolecular interactions that are imposed on TPA+occluded in the channel intersections of silicalite-1. Especially the splitting of the methyl carbon signal should be the direct reflection of the interactions between the methyl groups and the silicate framework in the straight and zig-zag channels of silicalite-1. The fast motion of the methylene group next to N resulted in low CP efficiency in the1H-13C CP NMR experiments (Fig. 3), which may be responsible for the weak signals (63.4 and 16.8 ppm ) at the initial stage (before 15 h). The motion of these groups became slow due to the interaction/confinement imposed by the inorganic framework at longer crystallization time and the enhanced CP efficiency led to the growth of the signals. These intermolecular inorganic-organic interactions are the keys to understanding how the geometry of the organic structure directing molecules give rise to the specific pore architectures.
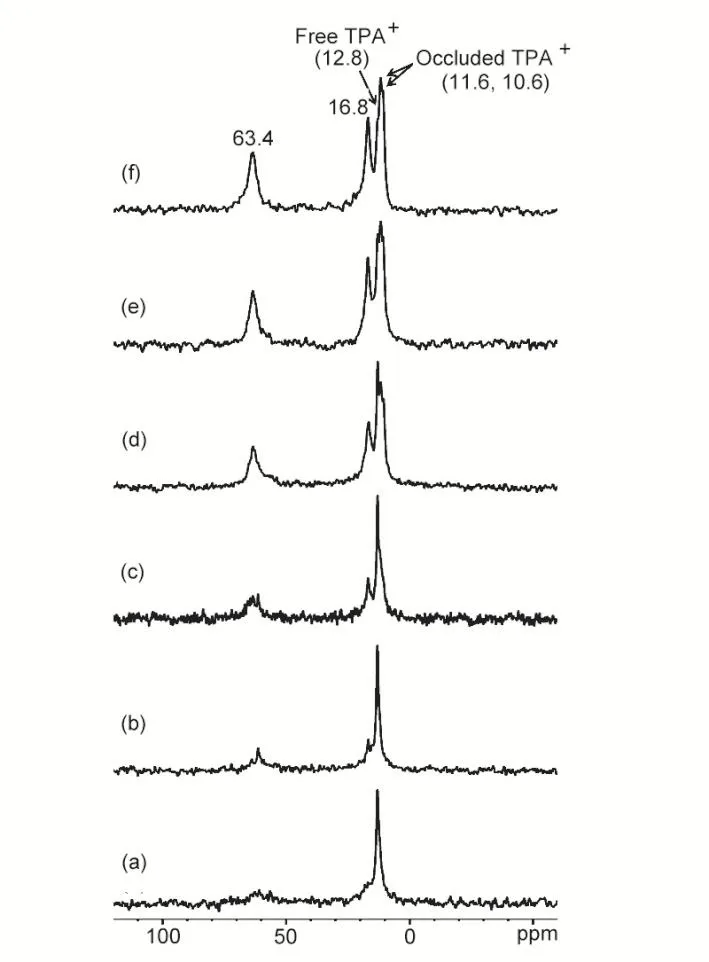
Fig. 3 1H-13C CP MAS NMR spectra of as-synthesized TPA-silicalite-1 samples heated at 180 °C for different time: (a) 0 h, (b) 5 h, (c) 15 h, (d) 16 h, (e) 18 h and (f) 20 h.
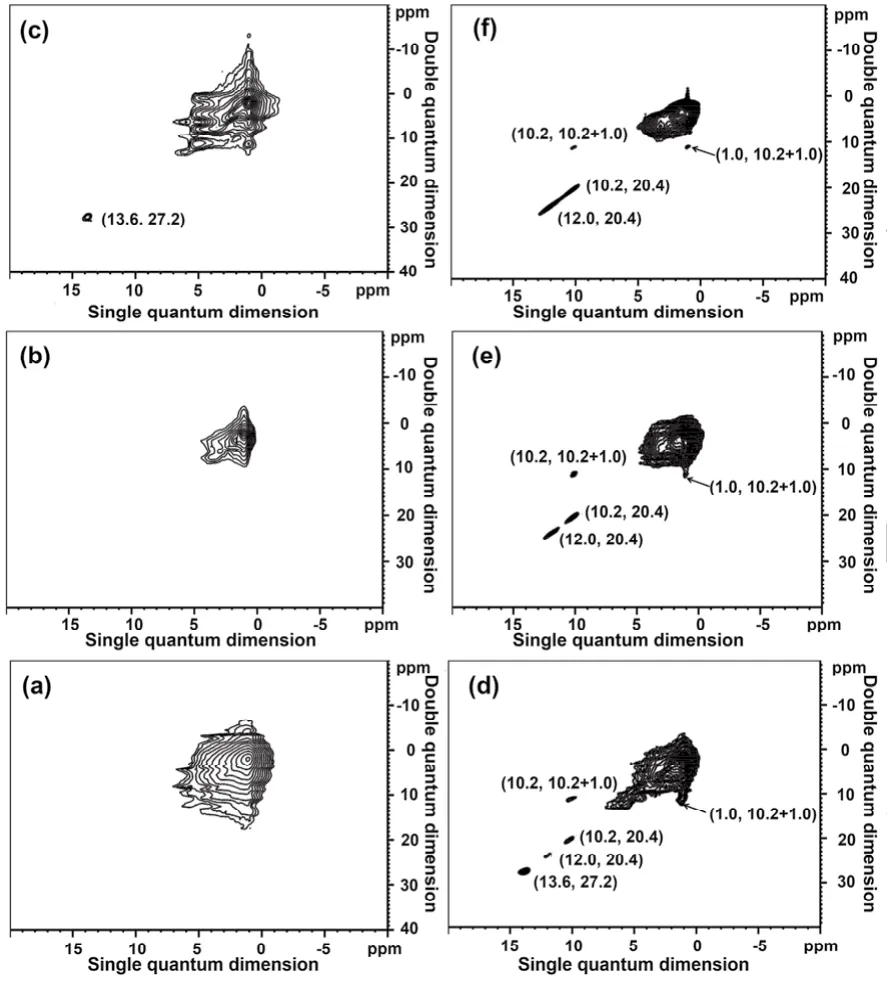
Fig. 4 1H DQ-SQ NMR spectra of as-synthesized TPA-silicalite-1 samples heated at 180 °C for different time: (a) 0 h, (b) 5 h, (c) 15 h,(d) 16 h, (e) 18 h and (f) 20 h.
Due to their favorable characteristics in studying local intermolecular interactions, 2D1H DQ-SQ MAS NMR techniques have been successfully employed to study the spatial correlation/interaction between the TPA+cations and the charged framework during the self-assembling process of zeolite ZSM-517,18, High-resolution 2D1H DQ-SQ NMR spectroscopy of dipolar-coupled spins in solids is an effective method to provide sufficiently selective structural information17. This technique makes use of the homonuclear dipole-dipole coupling between protons and takes advantage of the significant simplification of multi-spin dipolar-coupled networks under fast magic-angle spinning (MAS). In the 2D1H DQ-SQ spectrum, the spectral span of the DQ dimension is twice of that of the SQ dimension,i.e., the chemical shift of a correlation peak in the DQ dimension is the sum of the isotropic chemical shifts of the two same or different spins in the SQ dimension. Here, we used the 2D1H DQ-SQ MAS NMR to detect the spatial interactions between the TPA+cations and the silicates during the crystallization of silicalite-1. As shown in Fig. 4, for the samples heated less than 16 h (Fig. 4a–c), the auto-correlation (or diagonal)1H signals mainly come from the methyl (1.0 ppm), methylene (1.7 ppm)and methylene (3.2 ppm) adjacent to the nitrogen groups in the TPA+cations. Note that when the heating time increased to 15 h,a diagonal peak at (13.6, 27.2) ppm was observable as shown in Fig. 4c. Actually, this signal appeared after 11 h heating (Fig. S3(SI)). It can be attributed to the protons in SiO−··H ― OSi hydrogen bonds between lamellae and its appearance indicates that the SiO−··H―OSi are in close proximity one another in the amorphous phase. This type of hydrogen bonds is commonly observed in the intermediate phases of crystallized zeolites22,23.However, no cross-correlation (or off-diagonal) signals are observable for the SiO−··H ― OSi hydrogen bonds between lamellae, indicating that they are spatially away from the TPA+cations. Since the SiO−··H ― OSi hydrogen bonds between lamellae are detected at the introduction period, it is reasonable to assume that the SiO−··H ―OSi hydrogen bonds between lamellae may act as the connectors to assemble the inorganicorganic composites to form the intermediate layered silicates. In fact, layered silicates have been found to be co-phases in the synthesis of ZSM-5 and ZSM-1124,25. Recently, Lupulescu and Rimer identified a 2D layer nucleation growth process of ZSM-5 through time-resolved in situ AFM measurement by capturing growth progress pictures of ZSM-526. The crystallization process of silicalite-1 is an oriented 2D layer growth process, but until now no molecular-level evidence shows which one plays the connecting role in bridging the layers together. Here, the identified SiO−··H―OSi hydrogen bonds between lamellae at 13.6 ppm are most likely the connectors to bridge the neighboring polymerized silicates together.
When the heating time reaches 16 h (Fig. 4d), other two diagonal signals at (10.2, 20.4) ppm and (12.0, 24.0) ppm are observable and they also correspond to the SiO−··H ― OSi hydrogen bonds7. The SiO−··H―OSi hydrogen bond at 12.0 ppm may have similar structure property as the one at 10.2 ppm,but the different crystallographic sites in the charged framework lead to different chemical shifts. These two signals can be assigned to the SiO−··H―OSi hydrogen bonds in the [415262]cages. In order to avoid the confusion on the different SiO−··H―OSi hydrogen bonds, the SiO−··H―OSi hydrogen bonds at 10.2 and 12.0 ppm are defined as the SiO−··H―OSi hydrogen bonds in-cage. It is interesting to note that an off-diagonal peak pair at(10.2, 11.2) ppm and (1.0, 11.2) ppm appears at this stage,indicating that the protons in the SiO−··H―OSi hydrogen bonds in-cage are in close spatial proximity to those in the methyl groups of TPA+cations. However, the protons in the SiO−··H―OSi hydrogen bonds in-cage at (12.0, 24.0) ppm do not show any correlation with those of TPA+cations probably due to the long distance between them, but only the auto-correlation signal at(12.0, 24.0) ppm is visible. In addition, the auto-correlation signal at (13.6, 27.2) ppm is still present for the protons of the SiO−··H―OSi hydrogen bonds between lamellae. It is wellknown that framework defect sites are required in as-made highsilica zeolites for the charge compensation of cationic organic structure-directing agents. In Fig. 4d, 2D1H DQ-SQ MAS NMR results clearly demonstrate that the formation of SiO−··H―OSi defect sites in-cage is associated with the local charge balance between the positive SDA and the zeolite framework with negative charges during crystallization processes. After the heating time further increases to 18 h and 20 h (Fig. 4e, f), the off-diagonal signals at (10.2, 11.2) ppm and (1.0, 11.2) ppm are always present, thus they can be used as the characteristic signals to represent that the MFI channel systems have been fully established. The 2D1H DQ-SQ spectra clearly demonstrate the close geometric correlation between the pore architecture and occluded organic cations. The cross-correlation signals indicate that the SiO−··H―OSi hydrogen bonds in-cage are used as the stereoscopic negative charge centers to counterbalance the positive charges from TPA+cations, and the formed semi-crystal polymerized silicates with occluded TPA+cations, which do not show the long range periodic crystalline properties of XRD patterns in Fig. 1, can act as the crystal nucleus to promote the growth of periodical zeolite framework. Therefore, it can be concluded that the stereoscopic electrostatic interactions between the negative charge centers (SiO−··H―OSi hydrogen bonds in-cage) and the positive charge centers (TPA+), instead of the van der Waals interactions from the occluded organic molecules with the silicates, play the dominant structural direction role in forming these semi-crystal phases with MFI channel property. Furthermore, the stereoscopic electrostatic interactions between the charged framework and the occluded TPA+cations also facilitate the crystallization process of silicalite-1. Note that the diagonal signal at (13.6, 27.2) ppm from the SiO−··H ― OSi hydrogen bond between lamellae disappears with increasing the heating time (Fig. 4e and 4f). This observation confirms that the SiO−··H―OSi hydrogen bonds between lamellae only play the bridging role to connect the polymerized silicates together during the crystallization process,and they do not act as the negative charge compensating centers.
In order to further study the role of SiO−··H―OSi hydrogen bonds during the crystallization process of silicalite-1,2H MAS NMR experiments were performed on the as-synthesized silicalite-1 samples at different heating time. The strong signals from water and TPA+cations in1H MAS NMR spectra (Fig. S4(SI)) may cover the weak signals in the framework (Fig. S4),hence2H MAS NMR spectroscopy was used as an alternative technique to follow the crystallization process of silicalite-1. The advantage of using2H MAS NMR is that the magic-angle spinning can concentrate the2H powder patterns into tens of spinning sidebands and also remove the inhomogeneous firstorder quadrupole interaction. The highly resolved2H MAS NMR spectra match the critical requirement to study the zeolite crystallization process. As shown in Fig. 5, even though the quadrupolar coupling constant (QCC) is relatively small, the2H MAS NMR spectra spread over a wide range (~100 kHz) and it still needs quite a long time (about 5 days) to acquire a wellresolved spectrum. The qualitative information could be derived from the2H MAS NMR spectra by analysis of the isotopic chemical shifts (δiso) and QCC. Different signals from hydrogen bonds can be clearly identified in the spinning sidebands region in Fig. 5 (right panel). The sites with different chemical shifts can be directly observed and their quadrupole parameters can be reasonably determined by simulating the spinning sidebands.Fig. S5 (SI) depicts the simulated spectra of different2H sites,and Table S1 (SI) provides their NMR parameters.
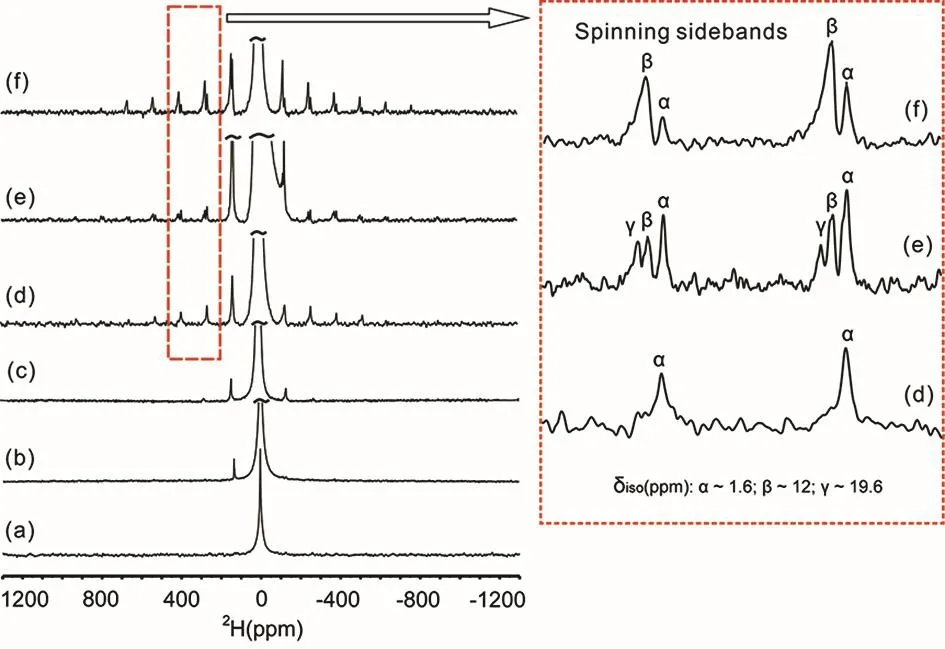
Fig. 5 2H MAS NMR spectra of as-synthesized TPA-silicalite-1 samples heated at 180 °C for different time: (a) 0 h, (b) 5 h, (c) 15 h,(d) 16 h, (e) 18 h and (f) 20 h.
At the beginning (Fig. 5a), only an intense narrow2H signal with δiso= 4.7 ppm and no spinning sidebands is observable,corresponding to water molecules with high mobility27. After heating the mixture of raw material for 5 h, first-order spinning sidebands extending over tens of kHz appear (Fig. 5b), implying that apart from the highly mobile water molecules, some water molecules are hydrogen-bonded with terminal oxygen atoms of silicates (δisois also 4.7 ppm)22and thus the mobility is largely restrained (leading to the large QCC). When the heating time is increased to 15 h, the spinning sidebands of the hydrogenbonded water molecules become intense, i.e., weak second-order spinning sidebands are visible (Fig. 5c), corresponding to a QCC of ca. 20 kHz (Table S1). The hydrophobic effect of TPA cations may make the polymerized silicates connected by these hydrogen-bond water molecules to form the clathrate-like ensemble of inorganic-organic composites. The inorganicorganic composites formed by the silicates and water molecules around the TPA+molecules separate the TPA+cations from each other, but the inorganic-organic composites do not have the structural properties of MFI channel intersections yet.
After 16 h heating,2Hαsignal with δiso(D) = 1.6 ppm and a QCC of ca. 120 kHz was observed (Fig. 5d). It is well-known that the signal at 1.4-1.6 ppm is due to the SiOH nests for the missing tetrahedral framework atoms (T vacancies) in zeolite framework27,28,29, which is schematized in Fig. S6 (SI). The large QCC and the wide-spread of spinning sidebands of the 1.6 ppm signal indicates that a hydrogen bonded network among four silanols may be formed. Because the proton signal from the SiOH nest is covered by the methyl groups of TPA+, this signal cannot be observed in the1H MAS spectrum in Fig. S4.Meanwhile the similarity of the surface dangling Si-OH groups and the SiOH nests (Fig. S6) causes difficulty in differentiating them by1H MAS NMR spectrum. It has been reported that SiOH nests are responsible for the poor stability of zeolites in hot liquid water30. Here our work shows that2H MAS NMR spectroscopy could be a nice choice to fix the dilemma of identifying the SiOH nests from the dangling Si-OH.
After the heating time increase to 18 and 20 h (Fig. 5e and 5f),the signal at 1.6 ppm is still observable, which indicates that the SiOH nests still exist in the framework, even though the longrange periodic zeolite structures have been fully established.Meanwhile, other two hydrogen-bonded hydroxyl species are clearly observed with the isotropic chemical shift at 12.0 and 19.6 ppm respectively (Fig. 5e). The2Hβsignal with δiso(D) =12.0 ppm can be assigned to the SiO−··H―OSi hydrogen bonds in-cage, while the2Hγ signal with δiso(D) =19.6 ppm can be assigned to the SiO−··H―OSi hydrogen bonds between lamellae bridging the siloxy (SiO-) groups from two silicates22,23. But the2H chemical shift of the SiO−··H―OSi hydrogen bonds between lamellae is higher than the corresponding1H signals of the layered silicates and those in the small cages, which often occur at 16–18 ppm and 10.2–11.6 ppm, respectively11,22. There is a linear relationship between the O−··H―O bond length and the proton chemical shift, and a shorter O−··H―O bond length corresponds to a higher proton chemical shift31. The O−··H―O hydrogen bond is a function of the following three quantities: the O―H covalent bond length, the O−··H hydrogen-bond length,and the O−··H―O bond angle. The higher δiso chemical shifts of SiO−··H―OSi defect sites (δiso(D) = 12.0 ppm vs. δiso(H) = 10.2 ppm) should be due to the H/D isotope effect which makes the O−··D bond distances shorter than the O−··H bond distances.However, the much higher δiso(D) value of SiO-··H ― OSi hydrogen bond between lamellae (δiso(D) = 19.6 ppm vs. δiso(H) =13.6 ppm) is not just due to the H/D isotope effect on the O−··H bond distances, but the near 180° O−··H―O bond angle, which is rare in crystals, also contributes to the large chemical shift. It is interesting to note that the2Hγsignal (19.6 ppm) is observable at 18 h, indicating that the SiO−··H― OSi hydrogen bonds between lamellae are still present at this stage, but they are not spatially close as reflected by the disappearance of the diagonal signal at (13.6, 27.2) ppm at the same time (Fig. 4). Above observations confirmed that the spatial proximity is the key to realizing the transformation from the SiO−··H―OSi hydrogen bond between lamellae to the SiO−··H―OSi hydrogen bonds incage and form the crystallines. The signal from the SiO−··H―OSi hydrogen bond between lamellae observed in2H MAS NMR corresponds to the non-fully crystallized materials during the crystallization, which is consistent with our XRD patterns.
In order to confirm that the formation of SiO−··H― OSi hydrogen bond between lamellae is not due to the specific synthesis method, silicalite-1 zeolite was also synthesized under the hydrothermal condition at 175 °C for 14 days. As shown by its XRD pattern (Fig. S7 (SI)), the sample is highly crystallized with MFI structure.1H MAS NMR spectrum of as-synthesized silicalite-1 in Fig. S8 (SI) shows a weak and broad signal at 16.0 ppm, which can be attributed to the SiO−··H―OSi hydrogen bond between lamellae. This observation is not unusual for silicalite-1 synthesized under hydrothermal condition, but the signal was not much discussed before because it is too weak and too broad to provide structural information. Together, our1H 2D DQ-SQ and2H MAS NMR results demonstrate that the SiO−··H―OSi hydrogen bonds between lamellae are formed during the crystallization process of silicalite-1 zeolites. The evolution of the SiO−··H ― OSi hydrogen bonds between lamellae during the crystallization indicates that they would serve as the connectors to bridge the silicates together and fuse themselves to form the long-range periodic framework.
The above NMR observations are helpful to understand how the inorganic-organic composites have been assembled together to form the zeolite frameworks. A schematic is presented in Fig.6 to describe the crystallization process of silicalite-1. In this proposed model, during the introduction period, the van der Waals interactions promote the formation of tight inorganicorganic composites; then at the nucleation and crystal growth stages, the semi-organized solid phase or silicalite-1 crystal nucleus is formed through the stereoscopic electrostatic interactions between the SiO−··H―OSi hydrogen bonds in-cage and the TPA+cations, which made the formed framework structures with the MFI stereoscopic characteristics, meanwhile,the SiO−··H―OSi hydrogen bonds between lamellae bridge the silicates together to realize the 2D layer growth process of silicalite-1; Consequently, the intermediate phases are finally transformed into the more stable periodic crystal phases with boundary morphology.
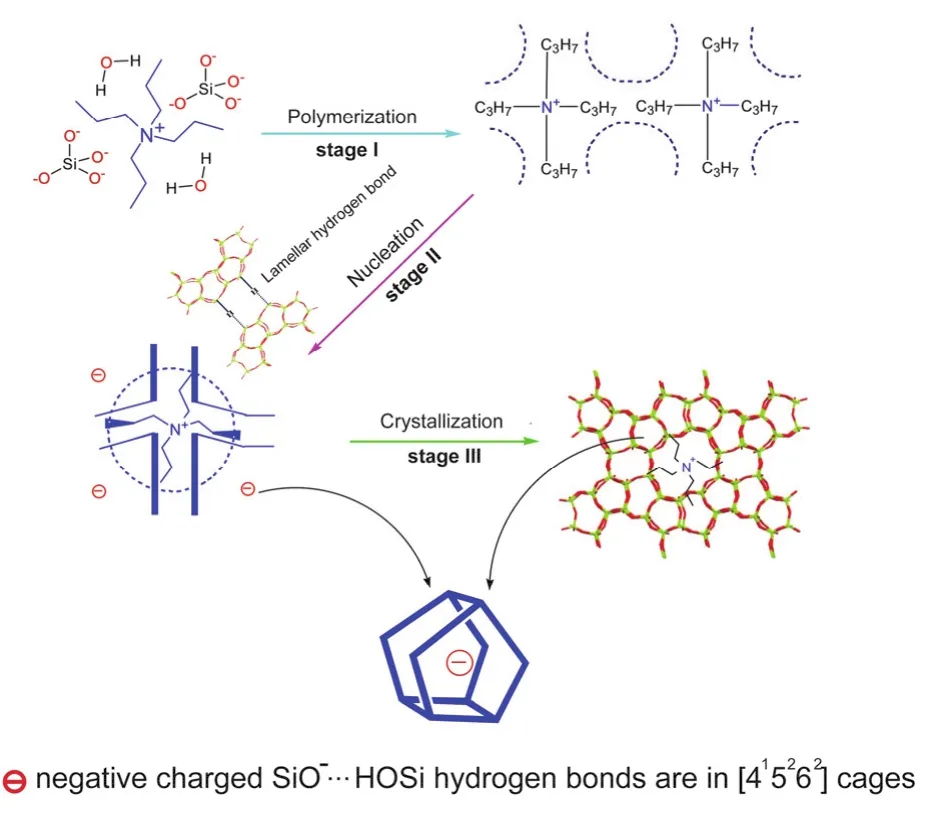
Fig. 6 A schematic of proposed crystallization mechanism of TPA+-mediated solvent-free synthesis of silicalite-1.
4 Conclusions
Our experimental results demonstrate that the hydrogen bonds play important role during the crystallization process of silicalite-1 zeolite. At the introduction period, the water hydrogen bonds and the SiO−··H ― OSi hydrogen bonds between lamellae organize the silicate species to form the inorganic-organic composites. During the nucleation and crystallization stage, the spatial proximity between the SiO−··H―OSi hydrogen bond between lamellae is the key to realize the transformation from the SiO−··H―OSi hydrogen bond between lamellae to the SiO−··H―OSi hydrogen bonds incage, and this transformation provides the negative charge centers for counterbalancing the positive charges from the TPA+cations, leading to the crystallization of amorphous phases. The stereoscopic electrostatic interactions organize the disordered inorganic-organic composites to form the silicalite-1 zeolite channel systems. This work provides detailed information for understanding the mechanism of zeolite formation, especially with respect to the local arrangement between SDA and the zeolite framework during nucleation and crystallization.
Supporting Information:available free of charge via the internet at http://www.whxb.pku.edu.cn.
