
几种三嗪基多哌嗪类二硫代卡巴腙衍生物的合成与表征
2020-07-13 03:26罗陈林蒋紫薇雷金玉朱嘉君蒋选丰
黄冈师范学院学报 2020年3期
吴 尧,罗陈林,蒋紫薇,雷金玉,朱嘉君,蒋选丰
(1.湖北大学 材料科学与工程学院,湖北 武汉 430000; 2.黄冈师范学院 化学化工学院,湖北 黄冈 438000)
硫代卡巴腙衍生物在荧光探针[1]、配位催化[2]、超分子自组装[3-6]以及高分子聚合[7-8]等方面有着广泛的应用。因此,通过化学合成方法实现不同功能的硫代卡巴腙衍生物的可控制备已经受到人们广泛关注[9]。从文献报道来看[10],常见的链状烷基胺取代的二硫代卡巴腙衍生物是通过原位反应“一锅法”合成得到的,但是所得化合物性质极不稳定,易与空气中的氧气反应形成过硫化物。因此,这类链状烷基胺取代的硫代羧酸衍生物很难实现分离与表征,并且难以在室温下存放,进而影响与金属离子的配位自组装以及硫-烯和硫-炔聚合物反应活性。为了改善脂肪胺基二硫代卡巴腙衍生物的化学稳定性,本课题组设计合成了一系列以1,3,5-位三嗪为中心结构的多取代哌嗪类二硫代卡巴腙化合物,这类新颖化合物中含有的哌嗪基团的半刚性六元环状骨架可以通过推-拉电子效应提高相应硫代卡巴腙化合物的化学稳定性[4]。本文报道的一系列含三嗪中心骨架的多哌嗪基二硫代卡巴腙衍生物具有独特的钳形结构,能和过渡金属离子形成配位作用强、理化性质稳定的超分子配合物,可应用于重金属离子荧光探针、OLED发光材料、VOCs荧光传感器和超分子光开关等方面。
1 试剂与仪器
三聚氯氰、1-Boc-哌嗪、四氢呋喃、二氧六环、二硫化碳、等均为分析纯。
1H NMR和13C NMR: Bruker AIVEN400(300MHz)和 Bruker AIVEN400(100MHz)(CDCl3或DMSO为溶剂,TMS为内标);质谱:BIFLEX III 型FT-ICR质谱仪(ESI)(CH3OH为溶剂)。
2 实验原理
根据文献报道的合成机理,三聚氯氰分子中的3个C-Cl单键的亲核取代活性有非常明显的差异。具体表现如下:通常三聚氯氰中1-位氯的亲核取代反应在低温下(-5 ~ 0℃)可以自发进行。然而,当反应温度升高到室温(不高于40℃),其中3-位氯的反应活性被激活,可以被亲核试剂取代,氯原子离去并生成1,3-位二取代产物。当温度升高到85℃到120℃区间,位于5-位的氯才能被取代,最终形成1,3,5-位三取代三嗪衍生物。很明显,三聚氯氰分子中不同位置的C-Cl键的分步断裂的活化能有较大差异。因此,本文通过改变反应温度将不同功能有机胺(哌嗪和烯丙基胺)分级引入到三嗪骨架上,得到一系列不同构型的N-杂环分子,最终通过与二硫化碳在KOH的碱液中反应生成具有特定配位和聚合功能的以三嗪为中心结构的对称和不非对称取代的哌嗪类二硫代卡巴腙产物5、9和12,并运用核磁共振谱仪和质谱仪对中间产物和目标分子进行了表征。
2.1 具有单一配位或者聚合功能基团的三嗪基多哌嗪类二硫代卡巴腙衍生物的制备
本文采用三聚氯氰和N-Boc哌嗪作为反应原料,以碳酸钾作为碱,在二氧六环溶液中加热至85℃,通过“一锅法”实现三聚氯氰中3个C-Cl键的断裂和亲核取代,生成中间体3。在第二步反应中,利用浓盐酸酸化脱保护生成中间体4[11]。以化合物4为原料,通过二硫化碳分子与哌嗪基团之间的亲核加成反应生成相应的二硫代卡巴腙产物(化合物5),如图1所示。

图1 化合物5的合成路径
2.2 具有多功能配位或聚合功能基团的非对称三嗪基多哌嗪类二硫代卡巴腙衍生物的制备
为了得到一系列不对称的双功能化哌嗪基二硫代卡巴腙衍生物,本文在化合物5合成方法的基础上进一步改进。原料三聚氯氰和N-Boc哌嗪(1∶2),在含有碳酸钾的二氧六环溶液中室温搅拌反应,成功实现三聚氯氰中1,3-位两个C-Cl键的断裂和亲核取代,生成中间体6。在第二步反应中,将4-吡啶基哌嗪与化合物6(1∶1)溶解在含有碳酸钾的二氧六环溶液中,同时升高反应温度至110 ℃,搅拌一天后得到化合物7。与化合物4的合成方法相同,利用浓盐酸酸化脱保护生成中间体8。与化合物5的合成方法不同,通过改变反应温度成功实现三嗪骨架中的2,4,6-位C-Cl键的可控断裂并分级亲核取代,从而引入不同的功能基团得到相应不对称功能分子。然后,通过二硫化碳分子与化合物8中哌嗪基团之间的亲核加成反应生成相应的二硫代卡巴腙产物9,具体合成步骤如图2所示。

图2 化合物9的合成路径
在上述分级合成方法基础上,将具有共价交联聚合特性的二烯丙基胺基团引入到三嗪骨架上,得到另外一类双功能三嗪基哌嗪二硫代羧酸模块分子11和12。具体合成方法如图3所示。
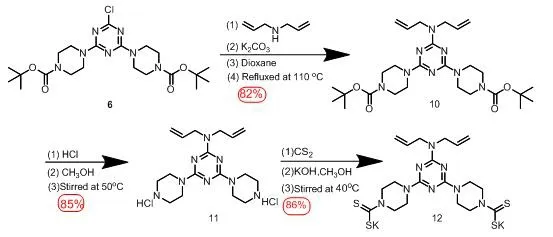
图3 化合物12的合成路径
3 合成与表征
3.1 化合物5(2,4,6-三(4-(1-二硫代羧酸基)哌嗪基)-1,3,5-三嗪钾盐)的合成制备与表征
化合物3 (2,4,6-三(1-Boc哌嗪基)-1,3,5-三嗪)的合成:将三聚氯氰(0.92 g,5.03 mmol)、 1-哌嗪-Boc(3.3 g,15.21 mmol)、 K2CO3(0.69 g,5 mmol)加入到80 mL四氢呋喃中,85℃搅拌回流,反应48 h以后经过真空旋干、水洗、过滤、烘干得到干燥的化合物3(2.70 g,收率85%)。1H NMR (300 MHz, Chloroform-d)δ3.73 (t,J=5.1 Hz, 11H), 3.44 (t,J=5.1 Hz, 12H), 1.48 (s, 27H);13C NMR(100 MHz, Chloroform-d)δ165.39, 154.80, 79.82, 67.06, 43.03, 28.41。
化合物4(2,4,6-三(4-哌嗪基)-1,3,5-三嗪)的合成:称取化合物3(2.80 g,4.42 mmol),以20 mL乙醇为溶剂,加入约5 mL水和5 mL浓盐酸(1 M),50 ℃搅拌回流,反应48 h。反应完成后将溶剂旋干,加入少量水,超声后逐滴加入NaOH浓溶液至混合物pH达到14,加入约50 mL二氯甲烷不断搅拌并萃取,将分液后的有机相备用,将水相使用50 mL二氯甲烷进行再次分液,重复以上萃取操作3~5次,将所有分液得到的有机相混合后使用约4 g无水硫酸钠干燥并减压旋干溶剂即得到化合物4(1.19 g,收率 81%),HR-ESI-MS (CH3OH) m/z:[M+H]+, 分子式为 C15H27N9+, 理论值为334.25; 实验值为 334.25。
化合物5的合成:以50 mL密封直形反应管为容器,将化合物4 (1.20 g,3.60 mmol)和KOH(0.5 g,8.93 mmol)溶于20 mL乙醇溶剂中通氮气下搅拌,升温至40 ℃反应过夜,密封条件下加入CS2(1 mL,15 mmol),继续反应24 h,反应完成后将溶剂旋干,加入3~5 mL甲醇,超声溶解,再灌入约10 mL乙醚重结晶得到淡黄色粉末状的目标化合物5 (1.94 g,收率80 %,纯度96%,熔点262~263 ℃)。1H NMR (300 MHz, DMSO-d6, 298 K)δ3.64 (s, 12H, ), 3.35 (s, 14H);13C NMR (100MHz, DMSO-d6)δ214.60, 165.12, 49.33, 43.21,其核磁共振氢谱与碳谱图分别见图4和图5。

图4 化合物5的核磁共振氢谱图

在目标化合物5结构中,其三嗪骨架中1,3,5-位由哌嗪二硫代羧酸基取代,是一类具有对称结构的分子。核磁共振谱1H NMR 和13C NMR 结果表明文中所合成的目标化合物5结构正确,并且该化合物在空气中可稳定存放,其化学稳定性较好。这种半刚性含有氮硫的三脚架配体能与各种过渡金属离子通过强配位作用形成功能超分子配合物组装体。
3.2 化合物9(2,4-二(1-二硫代羧酸基-4-哌嗪基)-6-(1-(4-吡啶基)-4-哌嗪基)-1,3, 5-三嗪钾盐)的合成制备与表征
化合物6 (2,4-二(1-Boc基-4-哌嗪基)-6-氯-1,3,5-三嗪)的合成:以三聚氯氰(1 g,5.46 mmol)、 1-哌嗪-Boc(1.03 g,5.50 mmol)、碳酸钾(3.8 g,27.5 mmol)为反应物,85℃搅拌回流,反应48 h以后经过真空旋干、水洗、过滤、烘干得到干燥的化合物6 (2.26 g,收率85 %)。1H NMR (300 MHz, Chloroform-d)δ3.82 (s, 8H), 3.51 (s, 8H), 1.53 (d,J=2.0 Hz, 18H);13C NMR (100 MHz, Chloroform-d)δ165.39, 154.80, 79.82, 43.03, 28.41;HR-ESI-MS (CH3OH) m/z:[M+H]+, 分子式为 C21H34ClN7O4+,理论值为 483.24; 实验值为 484.6。
化合物7(2,4-二(1-Boc基-4-哌嗪基)-6-(1-(4-吡啶基)-4-哌嗪基)-1,3,5-三嗪)的合成制备:以化合物6(2.30 g,4.77 mmol)、(4-吡啶基)-1-哌嗪(0.80 g 4.91 mol)、K2CO3(1.97 g,14.31 mol)为反应物,85℃搅拌回流,反应48 h以后经过真空旋干、水洗、过滤、烘干得到干燥的化合物7(2.47 g,收率 85%)。13C NMR (100MHz, Chloroform-d)δ165.35 (d,J=3.9 Hz), 154.81 , 149.33, 108.25, 79.91, 45.81, 42.74 (d,J=79.0 Hz), 28.42; HR-ESI-MS (CH3OH) m/z: [M+H]+, 分子式为C30H46N10O4+, 理论值为 611.37; 实验值为 611.38。
化合物8(2,4-二(1-Boc基-4-哌嗪基)-6-(1-(4-吡啶基)-4-哌嗪基)-1,3,5-三嗪)的合成制备:以化合物7(2.3 g,3.77 mmol)为反应物。反应完成后将溶剂旋干,加入少量水,超声后逐滴加入NaOH浓溶液至混合物pH达到14,加入约50 mL二氯甲烷不断搅拌并萃取,将分液后的有机相备用,将水相使用50 mL二氯甲烷进行再次分液,重复以上萃取操作3~5次,将所有分液得到的有机相混合后使用约4 g无水硫酸钠干燥并减压旋干溶剂即得到化合物8(1.24 g,收率 81 %)。1H NMR (300 MHz, DMSO-d6)δ14.02 (s,1H), 9.69 (s, 4H), 8.29 (t,J=5.4 Hz, 2H), 7.25 (dd,J=14.6, 7.0 Hz, 2H), 4.18~3.87 (m, 12H), 3.09 (s, 8H); ESI-MS (CH3OH) m/z: HR-ESI-MS (CH3OH) m/z:[M+H]+, 分子式为为C20H30N10+, 理论值为411.27;实验值为 411.27。
化合物9的合成:以化合物8 (1.3 g,2.04 mmol)和KOH (0.68 g,12.2 mmol)为反应物,40 ℃反应过夜,密封条件下加入 CS2(1 mL,15 mmol),继续反应24 h,反应完成后将溶剂旋干,加入3~5 mL甲醇,超声溶解,再灌入约10 mL乙醚重结晶得到白色粉末状的目标化合物9 (1.04 g,收率 80 %,纯度92%,熔点222~223 ℃)。1H NMR (300 MHz, DMSO-d6)δ8.17 (dd,J=6.0, 2.5 Hz, 2H), 6.83 (m, 2H), 4.38 (brs, 9H), 3.81 (t,J=4.1Hz,4H), 3.65(brs, 9H), 3.37 (d,J=5.6 Hz, 5H),具体核磁共振氢谱见图6。
在目标化合物9结构中,三嗪基2,4-位被(二硫代羧酸基)-哌嗪基取代,6-位被吡啶哌嗪基取代,结构左右对称,是一类新型的双功能氮杂环二硫代羧酸有机配体,可通过硫代羧酸中的两个硫原子与金(I)桥连配位形成一级超分子自组装体。接着,未参与一级组装的吡啶基团可继续与其它过渡金属进行二级配位组装,从而得到结构更加复杂并且功能更加多样的超分子聚集体。通过上述温度控制的分级取代反应,可以在三嗪骨架上的引入不同的功能基团,实现不对称功能模块分子的可控合成。

图6 化合物9的核磁共振氢谱图
3.3 化合物12 (2,4-二(1-二硫代羧酸基-4-哌嗪基)-6-(N,N-二烯丙基)-1,3,5-三嗪) 的合成制备与表征
化合物10(2,4-二(1-Boc基-4-哌嗪基)-6-(N,N-二烯丙基)-1,3,5-三嗪)的合成制备:以化合物6(1 g ,2.07 mmol)、二烯丙胺(1.3 mL,10 mmol)、K2CO3(1.38 g,10 mmol)为反应物,85 ℃搅拌回流,反应48 h以后经过真空旋干、水洗、过滤、烘干得到干燥的化合物10(0.93 g,收率82 %)。1H NMR(300 MHz, DMSO-d6)δ9.58 (s, 4H), 3.94 (t,J=5.1 Hz, 8H), 3.60 (dq,J=12.0, 6.7 Hz, 8H), 3.46 (q,J=7.0 Hz, 2H), 3.20~3.00 (m, 8H), 1.08 (t,J=7.0 Hz, 2H). 3.35 (s, 17H), 1.44 (s, 19H)。
化合物11 (2,4-二(1-氢-4-哌嗪基)-6-(N,N-二烯丙基)-1,3,5-三嗪盐酸盐) 的合成:以化合物10(0.9 g,1.65 mmol)为反应物。反应完成后将溶剂旋干,加入少量水,超声后逐滴加入NaOH浓溶液至混合物pH达到14,加入约50 mL二氯甲烷不断搅拌并萃取,将分液后的有机相备用,将水相使用50 mL二氯甲烷进行再次分液,重复以上萃取操作3~5次,将所有分液得到的有机相混合后使用约4 g无水硫酸钠干燥并减压旋干溶剂最后酸化即得到化合物11 (0.49 g,收率85 %)。
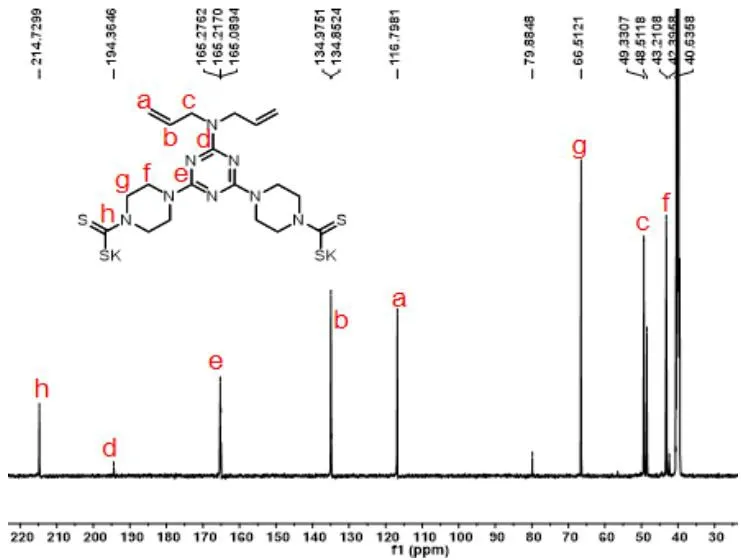
化合物12的合成:以化合物11 (0.49 g,1.41 mmol)和 KOH (0.4 g,7.10 mol)为反应物,40 ℃反应过夜,密封条件下加入 CS2(1 mL,15 mmol),继续反应24 h,反应完成后将溶剂旋干,加入3~5 mL甲醇,超声溶解,再灌入约10 mL乙醚重结晶得到白色粉末状的目标化合物12 (0.69 g,收率86 %,纯度96%,熔点205~206 ℃)。1H NMR (300 MHz, DMSO-d6)δ5.82 (brs, 2H),5.11 (t,J=13.7 Hz, 4H), 4.35 (t,J=5.3 Hz, 8H), 4.09 (d,J=5.7 Hz, 4H),3.62 (t,J=5.3 Hz, 8H);13C NMR (100 MHz, DMSO-d6)δ214.73, 194, 165.28, 165.09, 134.98, 116.80, 66.51, 49.33, 48.51, 43.21, 42.40,具体核磁共振氢谱与碳谱图见图7和图8。
在目标化合物12结构中,三嗪基1,3-位同样被(二硫代羧酸基)-哌嗪基取代。不同的是,5-位被烯丙胺基团取代,这类含双键的硫代羧酸配体可以在与通过配位作用与过渡金属离子组装成超分子聚集体后再利用硫-烯自由基聚合反应进行光诱导聚合,从而得到应用广泛的多维多孔高分子聚合物。

图7 化合物12的核磁共振氢谱图
本文提供了一种三嗪基多哌嗪类二硫代卡巴腙钾盐的制备方法,通过1H NMR,13C NMR和HR-ESI-MS等分析测试手段对上述化合物进行了表征,结果表明文中报道的中间化合物及目标产物结构正确。我们利用三聚氯氰中1,3,5-位3个C-Cl键亲核取代活性的差异,通过改变反应温度在其3个位置上实现不同官能团的分级引入,最终成功制备得到一系列稳定的三嗪基多哌嗪二硫代卡巴腙衍生物。这类化合物是一类具有强配位能力和共价交联活性的含硫有机配体和高分子聚合单体,可以通过配位作用与过渡金属离子组装成具有不同结构和功能的配位聚合物和超分子聚集体。除此之外,利用硫-烯和硫-炔聚合物等自由基聚合反应可以实现上述化合物在多维多孔高分子聚合物的可控光诱导聚合中的应用。基于此,本课题组正在开展相关后续研究工作。
猜你喜欢
农业工程学报(2022年13期)2022-10-09
中南大学学报(自然科学版)(2022年6期)2022-08-01
地球科学与环境学报(2020年2期)2020-03-26
鸭绿江(2018年12期)2018-12-28
分析化学(2016年11期)2017-04-25
现代计算机(2009年3期)2009-12-21
