
痣样基底细胞癌综合征1例并文献复习★
2020-07-17 00:44于洪岩高正君司小强谢沛霖林沛婷薛晓东
皮肤病与性病 2020年3期
于洪岩,高正君,司小强,谢沛霖,林沛婷,刘 琼,薛晓东
(1.西北民族大学医学院,甘肃 兰州 737000;2.甘肃省人民医院整形外科,甘肃 兰州 737000)
痣样基底细胞癌综合征(Nevoid basal cell carcinoma syndrome,NBCCS)又称Gorlin-Goltz综合征,是一种罕见的常染色体显性遗传性疾病,由皮肤基底细胞癌、多发性颌骨角化囊肿、掌角化障碍和足底凹陷、骨骼系统异常及多器官发育障碍组成的综合征。其临床表现多种多样,首诊原因各不相同,临床诊断较为困难,易误诊。国内外关于本病的报道极罕见。甘肃省人民医院整形外科收治1例NBCCS,现报告如下。
1 病例资料
1.1 一般情况 患者男,28岁,汉族,右耳前黑色素痣增大伴反复破溃半年余。2007年及2018年于我院行全身多处黑色素痣切除术,术后病检为基底细胞癌。2019年6月右耳前区无明显诱因出现黑色素痣,呈米粒样大小,表面粗糙,后增大至红豆样大小,伴反复破溃;颌下区、头顶部、左小腿胫前区、后背无明显诱因出现黑色素痣,无破溃。既往先天性唇腭裂,唇裂修补术后;先天性室间隔缺损,已行室间隔缺损修补术。追问其家族史,父亲有类似皮肤肿物生长史,2年前于我院确诊皮肤基底细胞癌,行恶性肿瘤扩大切除术,治愈出院。
1.2 专科检查 全身多处见散在色素痣,右耳屏前见直径15mm黑色素痣,突出皮面,表面凹凸不平,伴破溃,与周围组织界限清晰,基底无明显粘连。颌下区、头顶部、左小腿胫前区、后背等见无痛性黑色素痣,直径(7~13)mm,未见破溃。悬雍垂至硬腭部完全裂开,裂隙最宽处为1.5cm,裂隙两侧黏膜、肌层完整,口鼻腔相通,咽腔宽大,发音不清(图1a、b、c)。

图1
1.3 辅助检查 CT平扫示:大脑镰及小脑幕钙化(图2a、b);T2椎体半椎畸形,上段胸椎侧弯(图2c);腭裂(图2d),下颌骨多发骨囊肿(图2e)。血生化、血常规、肿瘤标志物、胸片等未见异常。
1.4 治疗 局麻下行右耳前区、颌下区、头顶部、左小腿胫前区、后背黑色素痣扩大切除术(皮损扩大切除 5mm)。
1.5 病理结果 基底细胞癌。
1.6 皮损组织病理 真皮浅层内有与表皮相连的基底样细胞团块,团块边缘见栅状基底样细胞增生,收缩间隙不明显,周边伴多数色素颗粒沉积。真皮浅中层见基底样肿瘤细胞团块浸润,周边排列成栅栏状,部分肿瘤细胞周边可见明显收缩间隙(见图3a、b)。
1.7 最终诊断 ① 痣样基底细胞癌综合征;② 先天性III度腭裂;③ 牙源性角化囊肿;④ 室间隔缺损修补术后;⑤ 先天性唇裂术后;⑥ 脊柱侧弯。
目前患者仍在随访中。
2 讨论
2.1 发病机制 NBCCS是罕见的常染色体显性遗传性疾病。本病由White[1]于1894首次描述。1960年由Gorlin和Goltz首次提出多基底细胞三联征,即基底细胞癌(Basal cell carcinoma,BCC)、牙源性角化囊肿(keratocystic odontogenic tumor,KCOT)、骨 骼 畸 形[2]。 发 病 率 1/256 000 ~ 1/57 000[3],男女比例1∶1,白种人发病率较高,有色人种较罕见[4]。NBCCS由多发性基底细胞癌、多发性颌骨角化囊肿、掌角化障碍和颅内异位钙化、面部畸形(畸形眼、唇腭裂、严重眼畸形)及骨骼系统异常、多器官发育障碍组成。NBCCS由PTCH1、PTCH2、SUFU等多种基因的显性突变引起[5],伴完全外显性和强变异表达能力。PTCH基因属抑癌基因,包括PTCH1和PTCH2。90% NBCCS由9号染色体长臂(q22.3-q31)中等位PTCH1基因突变引起,突变率约40%~80%[6]。李静远[7]将1个中国汉族NBCCS家系的致病基因定位在PTCH2基因染色体区域1p32.3,指出PTCH2是此家系最可能的致病基因。不同人种、不同家系PTCH基因突变和单核苷酸多态性分布存在差异,其具体突变机制需进一步研究。Hedgehog信号转导系统是NBCCS的重要信号传导通路,包括SHH、PTCH1、PTCH2、SMO、HIP及GLI家族。PTCH基因编码PTC蛋白。PTC蛋白为SHH的受体。正常情况下,SMO与PTC结合为无活性复合体。当SHH与PTC结合后,SMO与PTC复合体构型发生改变,SMO单独游离出而对下游转录因子GLI有正向调节作用,调节下游靶基因的转录。正常的PTCH基因可抑制SMO的活性从而抑制此信号通路向下游基因的信号传递。PTCH1或PTCH2基因突变并与SMO结合而持续激活hedgehog信号通路,促进了NBCCS的发展,引起一系列发育异常和肿瘤发生[8、9]。进行PTCH基因突变筛查可助于本病的诊断。PTCH基因突变在50%NBCCS患者中检测到,可作为一级诊断标准[10]。
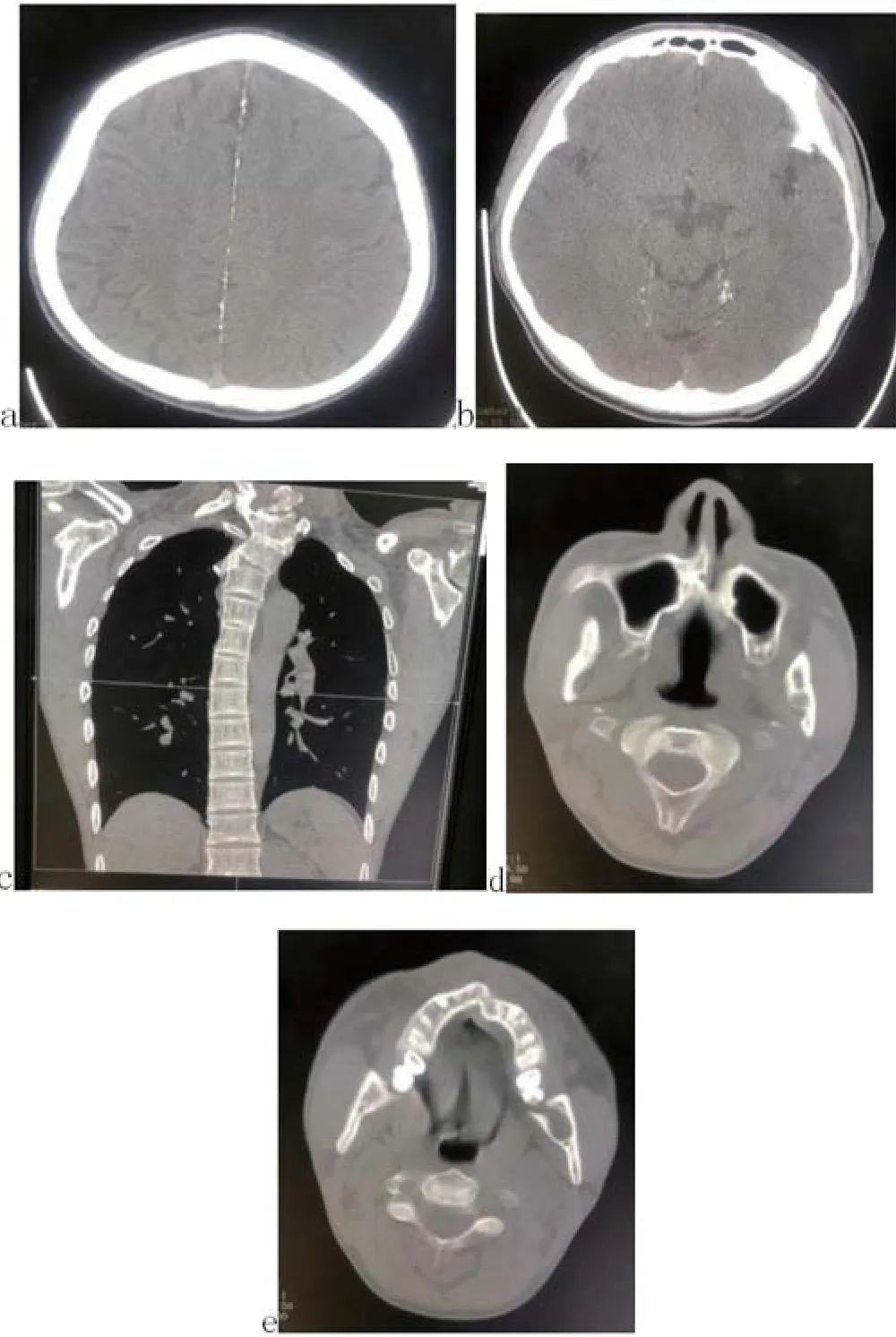
图2
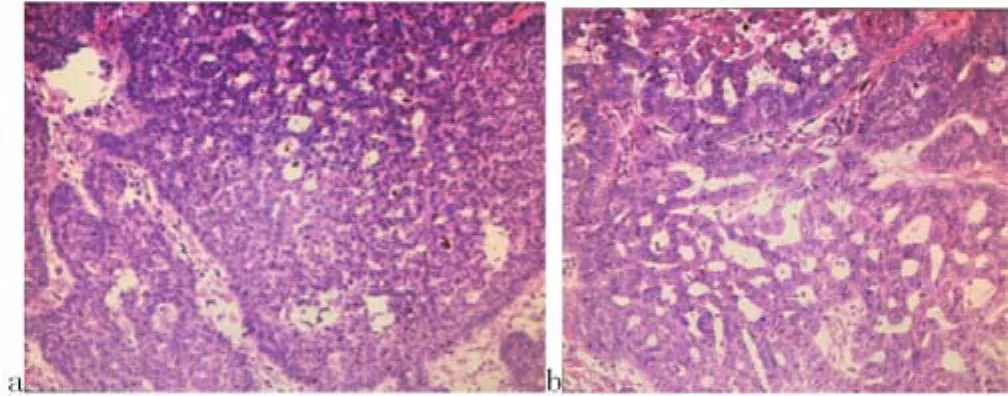
图 3 (HE 20×10)
2.2 诊断及鉴别诊断 临床诊断主要依赖诊断标准,PTCH基因检测可证实诊断。NBCCS诊断标准[11、12]为一级诊断标准,具体如下:① ≥2个基底细胞癌或1处发生在30岁之前,≥10个基底细胞痣或1处已存在20年以上;② 组织病理确诊的KCOT;③ ≥3个掌跖的坑状凹陷;④ 颅内多发异常钙化;⑤ 家族中第一级血亲为NBCCS。二级诊断标准:① 巨头畸形:枕额周长增大超过97%,额骨膨出;② 先天畸形,如唇腭裂、多或并指(趾)畸形、中重度的眶距增宽;③ 先天性骨骼发育异常;④ 脑髓母细胞瘤;⑤ 心脏或卵巢纤维瘤;⑥ 肠系膜囊肿。若符合2个一级诊断标准或符合1个一级诊断标准和2个二级诊断标准,则可诊断为NBCCS。本例患者符合一级标准中的①、②、④条及二级标准中的②、③条,故确诊为NBCCS。
本病临床特征繁多,为预防误诊漏诊,需明确鉴别诊断。皮损病理检查是“金标准”。BCC常与恶性黑色素瘤、皮肤鳞状细胞癌、脂溢性角化病等鉴别[13]。KCOT与成釉细胞瘤在影像上相似,病理可鉴别。与罕见综合征鉴别:① 副肿瘤性肢端角化症,好发中年,伴潜在肿瘤的肢端银屑病样皮损、基底细胞癌、稀毛、毛囊性皮肤萎缩、少汗;② 皮脂腺瘤伴内脏肿瘤综合征,常染色体显性遗传,好发中年男性,伴皮脂腺瘤、角化棘皮瘤、皮肤癌、结肠癌、十二指肠癌、子宫内膜癌等;③ 多发性丘疹性毛发上皮瘤,常染色体显性遗传,女性多见,好发面部鼻唇沟,呈光滑质硬结节。
2.3 治疗及预防 NBCCS涉及多个器官系统,治疗需多学科综合管理,定期复查CT、MRI等明确病变进展情况。NBCCS并不会明显改变患者预期寿命,若伴发脑髓母细胞瘤等恶性肿瘤则增加患者病死率。长期呈静止状态的基底细胞痣,一般不做特殊处理,应积极防晒,可用激光消融、光动力疗法、氟尿嘧啶及咪喹莫特药物治疗;若出现局部侵袭现象,应及时行根治手术。口服维A酸可对新发BCC起预防作用。口服vismodegib是抑制Hedgehog信号通路的药剂,使BCC减少了20倍[14],国内尚未用于临床。KCOT可根据病变具体情况采取刮除术或根治术,并联合电灼、化学腐蚀剂或冷冻疗法、自体骨移植来处理骨创,降低复发率。NBCCS应强制性进行遗传咨询,通过羊膜穿刺术或绒毛取样获得胎儿DNA进行产前诊断,减少患儿出生率。
猜你喜欢
医学美学美容(2022年12期)2022-10-17
数学学习与研究(2021年18期)2021-08-06
皮肤病与性病(2021年3期)2021-07-30
科学导报·学术(2019年11期)2019-09-10
婚姻与家庭·婚姻情感版(2019年6期)2019-06-03
东方教育(2017年14期)2017-09-25
中国科技纵横(2016年15期)2016-12-29
甘肃教育(2016年22期)2016-12-20
课程教育研究·下(2016年3期)2016-04-19
课程教育研究·学法教法研究(2016年1期)2016-03-17
