
HPLC-MS法测定黄芪中黄酮类成分的含量
2020-09-14 08:00陈春茗马丹凤
广州化工 2020年17期
陈春茗,龚 姗,陈 蕾,马丹凤
(1 湖南中医药大学第一附属医院,湖南 长沙 410007;2 湖南省儿童医院,湖南 长沙 410007)
黄芪为豆科植物蒙古黄芪或膜荚黄芪的干燥根,具有补气固表、利尿消肿、敛疮生肌等功效,在表虚自汗、阴虚盗汗、阳气虚弱、肺气虚、中气下陷、食少便溏、子宫脱垂等症上有较高药用价值[1-2]。现代药理研究表明,黄芪含有皂苷类、黄酮及多糖类成分,具有增强免疫功能、调节血压、抗菌、抗病毒、清除氧自由基、保护血管内皮功能、保肝、抗心肌缺血等多种药理作用[3-4]。黄芪药材中可分离出几十余种黄酮类成分,常见的有槲皮素、毛蕊异黄酮苷、芒柄花苷、毛蕊异黄酮、芒柄花素、染料木苷等[5-6]。本研究采用HPLC-MS法测定了黄芪药材中毛蕊异黄酮苷、芒柄花苷、毛蕊异黄酮、芒柄花素等4种含量较高的黄酮类成分含量,以期为黄芪药材质量控制提供科学依据。
1 仪器与试药
1.1 仪 器
API4000型三重四级杆质谱,美国应用生物系统;1100型液相色谱仪,美国安捷伦公司;AL204型电子分析天平,瑞士梅特勒-托利多公司;DL-360D超声波清洗器,上海之信仪器有限公司。
1.2 试 药
毛蕊异黄酮苷对照品(批号:15111012)、芒柄花苷对照品(批号:15041223)、毛蕊异黄酮对照品(批号:15090324)、芒柄花素(批号:15080717)均购买自广州牌牌生物科技有限公司,纯度均>98%;乙腈、甲醇、甲酸均为色谱纯,购自美国迈瑞达科技有限公司;水为去离子水。8批黄芪药材来源于不同产地,经笔者鉴定均为蒙古黄芪,见表1。

表1 黄芪药材来源与品种
2 实验方法
2.1 色谱条件
色谱柱:Acclaim 120 C18,戴安公司,2.1×100 mm,5 μm;流动相:乙腈(A)-0.3%甲酸溶液(B),梯度洗脱(0~10 min,18%~20% A;10~35 min,20%~24% A;35~52 min,24%~27% A;52~60 min,27%~34% A;60~65 min,34%~40% A;65~70 min,40%~50% A);流速:1.0 mL/min;柱温:检测波长254 nm,进样量:20 μL,柱温25 ℃。HPLC图谱如图1所示。
2.2 质谱条件
离子模式:电喷雾离子源(ESI);喷雾电压:241 kV;离子源温度:350 ℃;以氮气为干燥气,流速为1.5 L/min。扫描模式为选择离子监测(SIM),定量分析监测离子有毛蕊异黄酮苷m/z 445.11[M-H]-、芒柄花苷m/z 429.12 [M-H]-、毛蕊异黄酮m/z 283.06[M-H]-、芒柄花素m/z 267.07[M-H]-。
2.3 溶液的制备
2.3.1 混合对照品溶液
精密称定各待测成分对照品适量,加甲醇溶解,摇匀,制成混合对照品溶液,毛蕊异黄酮苷、芒柄花苷、毛蕊异黄酮、芒柄花素质量浓度分别为0.22、0.13、0.07、0.05 mg/mL。
2.3.2 供试品溶液
取样品适量,粉碎(过4号筛),精密称定1.0 g,加甲醇100 mL,回流提取1.5 h,减压浓缩,转移至5 mL量瓶,以甲醇定容至刻度,充分摇匀,再用0.22 μm微孔滤膜滤过,即得。
3 结果与讨论
3.1 系统适用性实验
取“2.3.1”项下配制混合对照品溶液适量,进样测定(按“2.1”项色谱条件),得到HPLC图谱,见图1。结果显示,毛蕊异黄酮苷、芒柄花苷、毛蕊异黄酮、芒柄花素保留时间分别为9.24 min、26.08 min、38.56 min、65.73 min,各成分均达到基线分离,峰形良好,理论板数均>1.5。
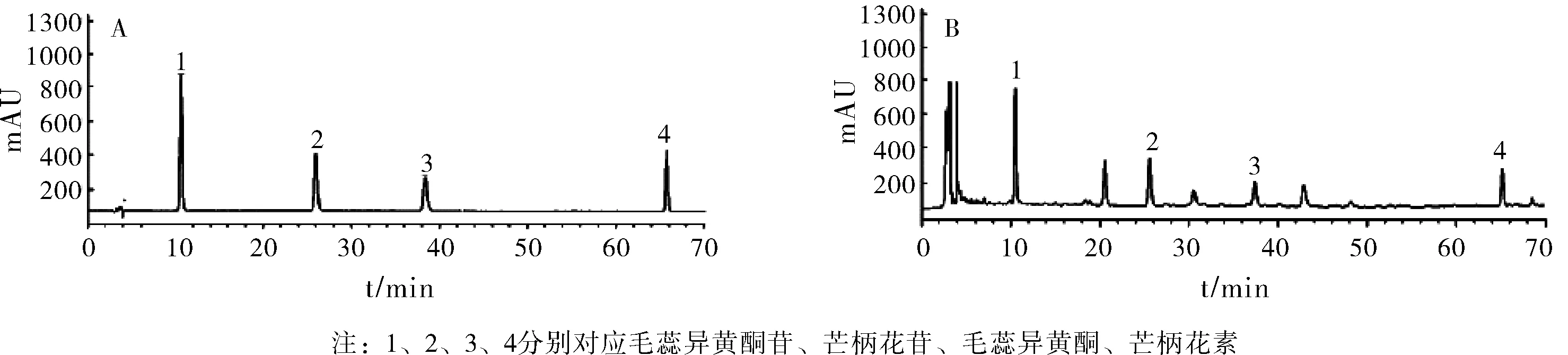
图1 混合对照品(A)和供试品(B)的HPLC图谱
3.2 线性关系考察
分别精密量取“2.3.1”项下混合对照品溶液1、5、10、50 μL,进样测定(按“2.1”项色谱条件),记录峰面积。以待测成分质量浓度(x,mg/mL)为横坐标,以峰面积(y)为纵坐标,做线性回归,得回归方程、线性范围,见2。

表2 回归方程及线性范围
3.3 精密度实验
取“2.3.1”项下配置的混合对照品溶液适量,进样测定(按“2.1”项色谱条件),连续进行6次测定,记录峰面积并分析,结果显示,毛蕊异黄酮苷RSD为0.23%,芒柄花苷RSD为0.56%,毛蕊异黄酮RSD为0.37%,芒柄花素RSD为0.18%,说明该方法具有良好精密度。
3.4 稳定性实验
取“2.3.2”项下配制的供试品溶液适量,室温下放置,并分别于0、4、8、12、18、24 h进行测定(按“2.1”项色谱条件),分析峰面积并分析,结果显示,毛蕊异黄酮苷RSD为0.15%,芒柄花苷RSD为0.27%,毛蕊异黄酮RSD为0.41%,芒柄花素RSD为0.22%,说明供试品溶液在室温下放置24 h具有良好稳定性。
3.5 重复性实验
配制供试品溶液(按“2.3.2”项相关方法),共制备6份,进样测定,色谱条件按“2.1”项,记录峰面积并进行含量测定。结果显示,毛蕊异黄酮苷含量为0.4212%,RSD为0.0842%;芒柄花苷含量为0.5187%,RSD为0.7923%;毛蕊异黄酮含量为0.2269%,RSD为0.0768%;芒柄花素含量为0.6582%,RSD为0.9228%;说明该方法具有良好重复性。
3.6 加样回收率实验
取已知含量的样品约0.5 g,共6份,并分别加入一定量待测成分对照品,配制供试品溶液(按“2.3.2”项下方法),进样测定(按“2.1”项下色谱条件),计算加样回收率,见表3。
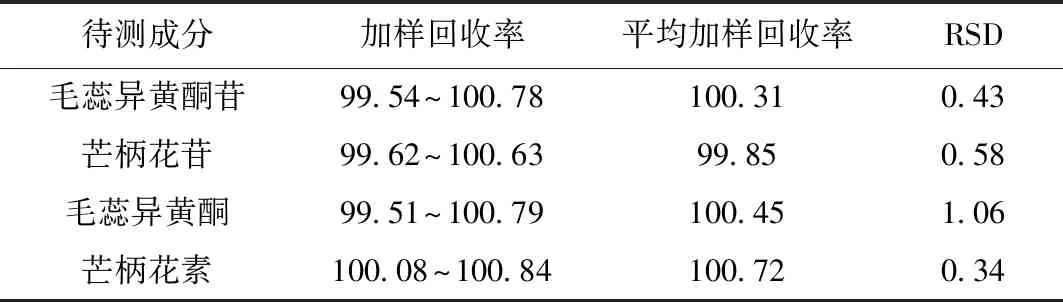
表3 加样回收率试验(%)
3.7 样品测定
8批黄芪药材,各取适量,配制供试品溶液(按“2.3.2”项下方法),进行测定(按“2.1”项下色谱条件),记录峰面积,计算各黄酮成分含量。见表4。
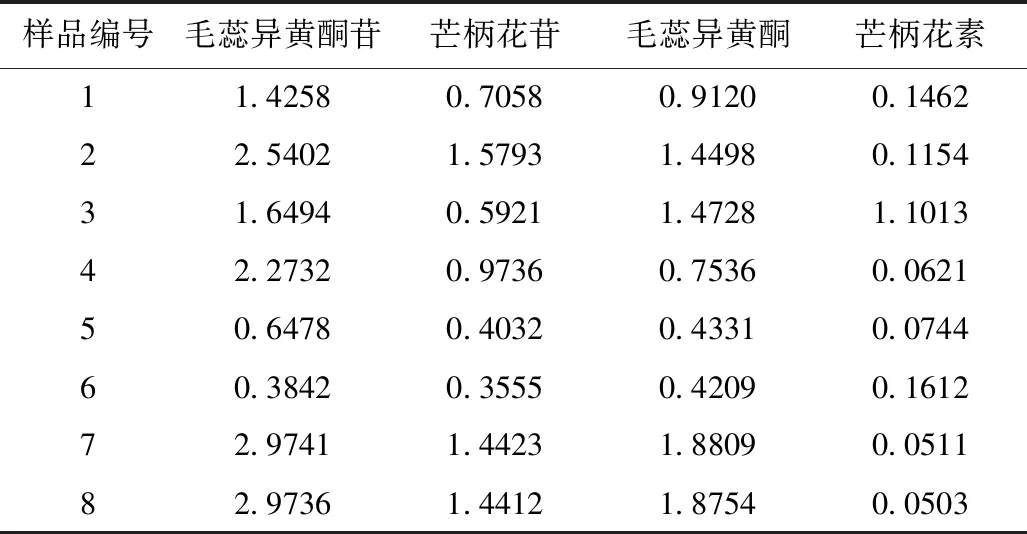
表4 黄芪药材中黄酮成分含量测定结果(n=3,mg/g)
3.8 检测方法选择
在前期条件摸索过程中,尝试以HPLC-DAD法测定黄芪中黄酮类成分含量,发现黄芪中芒柄花苷与毛蕊异黄酮等成分相互干扰不能实现完全分离。而HPLC-MS法选择性及灵敏度均较好,同时不要求待测成分达到完全分离,这对分析含有多种有效成分的中药有着明显优势[7],因此选择HPLC-MS法测定黄芪药材中黄酮成分含量。本研究应用的SIM模式较多反应监测模式专属性要低,使得一些待测成分在获取的离子流图中可出现多个色谱峰。对此,试验中测定了成分色谱峰的保留时间,以解决这一不足。
3.9 质谱条件优化
在待测成分的质谱条件选择优化过程中发现,毛蕊异黄酮苷、芒柄花苷、毛蕊异黄酮、芒柄花素等负离子检测模式下响应较好,故而选择负离子模式对黄芪中药中上述四种黄酮成分含量进行测定。
3.10 流动相考察
黄芪中黄酮类成分多呈弱酸性,因此应选择酸性缓冲系统[8],本研究分别对乙腈-磷酸、乙腈-甲酸系统进行了考察,发现抑制剂选用甲酸,黄酮各成分能够得到较好分离。同时还评估多种梯度流动相配比,结果发现,以乙腈(A)-0.3%甲酸溶液(B)为流动相系统,梯度洗脱(0~10 min,18%~20% A;10~35 min,20%~24% A;35~52 min,24%~27% A;52~60 min,27%~34% A;60~65 min,34%~40% A;65~70 min,40%~50% A)时4种黄酮类成分分离效果最好,色谱峰间分离度均在1.5以上,因而选择乙腈-0.3%甲酸溶液为流动相。
3.11 提取方法选择
有报道指出,在黄芪总黄酮提取上,连续回流提取法较超声法有着节省溶剂、操作迅速、获取有效成分高等多种优势[9]。本研究观察评估了超声法、冷浸法、回流法等三种提取方法,发现回流提取明显优于其他两种提取方法。此外,本研究还就不同提取时间进行了考察,发现回流1.5 h后,随着回流继续进行,毛蕊异黄酮、芒柄花素的峰面积逐渐减小,提示上述两种黄酮成分可能具有热不稳定性。故选择回流提取1.5 h进行黄芪中黄酮成分的提取。
3.12 检测波长选择
有研究显示,毛蕊异黄酮苷与毛蕊异黄酮的最大吸收波长为258 nm,而肩缝处于288 nm波长;芒柄花苷、芒柄花素最大吸收波长为248 nm,而肩峰处于290 nm[10-11]。采用二极管阵列检测器全波长对供试品溶液进行扫描,获取指纹图谱,以尽可能涵盖所有吸收峰为原则,发现各黄酮类成分在254 nm时,峰形及峰面积较好,因此选择254 nm为检测波长。
4 结 论
本研究建立HPLC-MS法同时测定黄芪中毛蕊异黄酮苷、芒柄花苷、毛蕊异黄酮、芒柄花素等黄酮类成分,具有较高的精密度、稳定性、重复性,且该法操作简便、迅速,可作为黄芪药材中黄酮类成分含量测定的质控手段。
猜你喜欢
世界科学技术-中医药现代化(2021年12期)2021-04-19
食品工业科技(2020年21期)2020-11-19
世界最新医学信息文摘(2020年34期)2020-05-23
中成药(2018年10期)2018-10-26
中成药(2018年9期)2018-10-09
天然产物研究与开发(2018年5期)2018-06-13
中山大学学报(自然科学版)(中英文)(2017年6期)2017-12-22
中成药(2017年6期)2017-06-13
云南中医学院学报(2015年2期)2015-07-31
中医研究(2014年4期)2014-03-11
