
RobustDEA:一种快速鲁棒的RNA-Seq数据寻找差异表达基因方法
2021-03-04 08:15王嘉瑞吴东洋
江苏科技大学学报(自然科学版) 2021年6期
张 礼, 王嘉瑞, 吴东洋
(南京林业大学 信息科学与技术学院,南京 210016)
下一代高通量DNA测序技术已经得到快速发展和普及,转录组测序技术(RNA-Sequencing,RNA-Seq)具有高通量,高信噪比,可重复性好,所需样本少等特点,被广泛应用的转录组学的各个研究领域中[1].寻找差异基因表达分析作为RNA-Seq数据分析中最基本应用之一,其目的是在不同对照组和控制组样本中,比如正常状态和药物治疗,健康和疾病个体,不同生物组织或者不同发育阶段等,识别出具有差异表达的基因.从RNA-Seq测序实验中获得海量读段数据后,首先需要将读段数据定位到参考基因组或者转录组上,从而统计出每个基因或者外显子上的读段数目.一旦获得基因或者外显子的读段数目,就可以进行差异表达分析.尽管RNA-Seq技术相比传统基因芯片具有很多优势,但是由于RNA-Seq测序本身技术上限制产生了很多困难,比如多源读段映射,测序偏好和小样本等问题,导致差异表达分析仍然是一个极具挑战的任务.
针对寻找差异表达基因所面临的种种挑战,提出了大量统计方法,通常可以分为两类:基于读段数目的方法和基于表达水平的两步法.基于读段数目的方法是比较在不同样本条件中基因上映射的读段数目.由于在不同样本之间,基因上读段数目变异性较大,若采用泊松分布会导致读段分布存在过散步(overdispersion)问题.因此,基于读段数目方法通常采用负二项(Negative binomial)分布,比如DESeq[2],DESeq2[3],baySeq[4],edgeR[5]等.相比泊松分布的单参数,负二项分布增加一个参数,能单独描述数据方差特性,从而能解决读段数据的过散步问题.此外,Voom使用正太线性模型来拟合读段数据,并且嵌入了数据分布的均值和方差的相关性信息[6].SAMSeq[7],NOISeq[8]以及NPEBSeq[9]等非参数模型,无需假设基因读段分布服从任何概率分布.但这类方法都忽略了读段的多源映射和数据偏差等问题,因此提出了基于表达水平的两步法:第一步利用现有表达水平估计方法计算出基因的表达水平,比如Cufflinks[10],PGSeq[11]等,这些方法在计算过程中考虑了读段多源映射和数据偏差等问题,从而获得更为准确的基因表达水平;第二步是利用获得的基因表达水平进行差异分析,比如CuffDiff2与Cufflinks,BDSeq[12]与PGSeq.由于考虑了更多的数据特征,基于表达水平的两步法通常能获得较好的结果.但由于两步法需要预先计算基因表达水平,其时间效率要比基于读段数目的方法低很多.因此,在面向寻找差异表达基因的单一任务时,目前主流做法是选择基于读段数目的方法,在较短的时间内获得较高的准确度[13].
针对RNA-Seq数据分析发现,同一个数据集中的基因,采用不同的寻找差异表达基因方法中会得到差异较大的结果[14].采用DESeq,baySeq,edgeR和Voom 4个方法来寻找MAQC数据集中的差异表达基因,表1展示了两个基因在不同方法中的P值和差异重要性排序,说明不同方法在分析同一个数据集是存在不一致性.导致这种现象除了数据集本身的生物特异性差异,以及不同研究者发表的基因表达数据的质量差异外,不同模型的假设不同或特点数据集偏好性,如NOISeq比较适合小样本数据集[15].此外,不同方法的结果报告格式不一致通常会导致结果理解和推断的困难.

表1 不同方法下基因载MAQC数据集上的结果
为了克服上述的问题,一种潜在可行方法是将当前算法结合起来,以获得更鲁棒的差异基因列表,其特点在于具有更高的统计能力,更少的假阳性和假阴性差异基因.研究人员通常是结合不同方法在同一个数据集上分析结果,从而推断出更稳定的结论.IDEAMEX是一个在线差异表达基因检测软件[16],该软件首先预设阈值得到每个方法所识别出的差异表达基因,在采用简单的投票方式选出4个方法都公认差异表达基因列表.consensusDE[17]和MultiRankSeq[13]方法都采用了基因排名求和的思想.首先不同方法根据p-value值大小对基因分别进行排序,然后将基因在不同方法中的排名求和,从而得到基因最终排名序列,排名越靠前的基因其越差异表达的可能性就越大.不同之处在于,MultiRankSeq方法结合了DESeq,edgeR和baySeq 3个方法,数据验证显示,DESeq和edgeR的差异基因集很接近,而baySeq找到的差异基因却和上述两个方法重叠的部分较少.图1是4个方法前1 000个最有可能的差异基因的Venn图,可以发现baySeq方法与其他3个方法的重叠基因很少,这将导致MultiRankSeq的结果易受到baySeq的影响.因此consensusDE方法采用了DESeq2,edgeR和Voom 3个方法,并在数据预处理步骤考虑消除不变基因的影响.上述方法通过投票或者排名求和的结合方式,找到被所结合方法共同认为最有可能的差异基因,但这些方法容易受到结合方法中单个方法的影响,反而降低了方法的准确性和鲁棒性.为了减轻单个方法对融合结果的影响,PANDORA方法对DESeq,edgeR,Vomm,NBPSeq,NOISeq和baySeq 6个方法进行加权融合[18].但是PANDORA方法需要预先产生与测试数据集特征相近的模拟数据集,通过模拟数据集计算出每个方法的权重值,此过程极其耗时.此外,PANDORA和MltiRankSeq都采用了DESeq,而不是性能更优越的DESeq2.
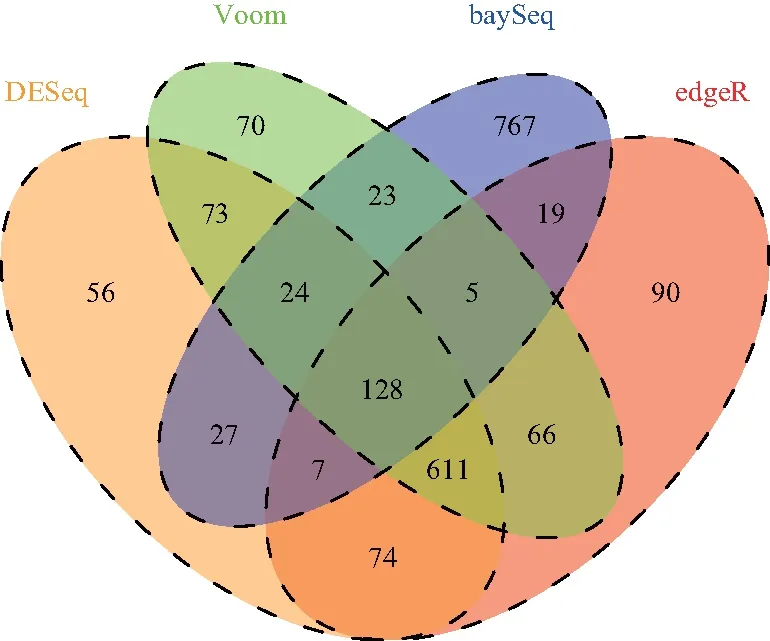
图1 DESeq, edgeR, Voom和baySeq
针对上述方法存在的问题,文中提出了一种鲁棒的寻找差异表达基因表达方法(Robust differential expression analysis based on RNA-Seq data,RobustDEA).RobustDEA方法选择了不同数据集中表现较好且稳定的4个方法:DESeq2, Voom, edgeR和NOISeq,通过加权方式结合多个寻找差异表达基因方法,其权值可快速从数据集中学习获到,并能有效的反映数据集的特点,从而使得RobustDEA方法可以在不同数据集上都能获得更稳定的结果.通过多个真实RNA-Seq测序数据集的验证表明,RobustDEA方法能在不同数据集上获得更加鲁棒的差异基因集.
1 方法
1.1 常见结合方法
基因芯片数据和RNA-Seq数据的差异表达基因分析中,多种结合方法被广泛应用到对不同方法检测结果的融合.目前,应用最为广泛的两种常规统计方法和专门针对RNA-Seq数据的最流行 PANDORA方法.首先,假设基因i使用寻找差异表达基因方法j获得其对应的P值pij,然后可以使用下面几种结合方法来进行不同方法结果的融合.
1.1.1 Fisher方法
根据Fisher方法,基因i通过m个方法估计的P值合并为:
(1)
合并后得到的统计量符合自由度为2m的卡方分布.χ2值越大,P值就越小,表示对应的基因其差异性就越明显.但是Fisher方法设计目的是合并具有相同零假设的统计检验方法在不同数据集中产生的P值,并不是合并同一个数据集上由不同统计检验方法产生的P值.
1.2.2 Stouffer方法
针对每个基因,标准的Stouffer方法首先将pj值转换为Zj=Φ-1(1-pj),其中Φ是标准正态分布的累积分布函数,每个基因的Z值为:
(2)
根据Z值大小对基因差异表达排序.标准Stouffer方法的优点易于为不同方法赋予不同权重值,因此提出改进的加权Stouffer方法:
(3)
式中:ωj为方法j的权重,其值可根据具体问题来选择,比如数据集的样本规模等.Z值越大,对应P值就越小,表示该基因的差异性就越显著.Stouffer方法和Fisher方法密切相关,其设计目的的假设都是合并具有相同零假设的统计检验方法在不同数据集中产生的P值.但加权Stouffer方法能便于对不同方法进行加权,而Fisher方法的卡方统计量,存在加权困难问题.
1.2.3 PANDORA方法
PANDORA方法的模型为:
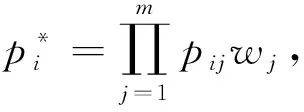
(4)
式中:ωj为方法j的权重,其值自动从模拟数据集中获得[18].此模拟数据集是基于正研究的数据集而产生,因此其权值能更为贴近研究的数据集.但模拟数据集的产生极其耗时,与结合方法的个数,测序样本数,基因个数等诸多因素有直接关系.
1.2 RobustDEA方法
针对不同寻找差异表达基因方法检测得到的差异基因集存在不一致情况,基于统计学中元分析的思想,文中提出一个快速鲁棒的RNA-Seq数据寻找差异表达基因方法RobustDEA,结合多个不同的差异表达基因方法,其模型为:
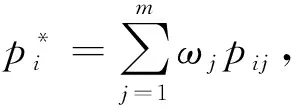
(5)
式中:ωj为方法j的权重值.RobustDEA方法采用自动加权方式,其权重值从数据集中自动计算得到.结合的每个方法先分别对数据集进行分析,获得每个基因的P值,可计算出每个方法在此数据集上的接收者操作特征曲线(receiver operating characteristic curve,ROC)下的面积(area under curve,AUC)值,从而自动估计出每个方法所对应的权重值为:
(6)
式中:AUCj为方法j的AUC值.通过自动加权方式,可以获得与当前分析数据集最合适的权重值,有利于提高RobustDEA方法的精确度和鲁棒性.
在RNA-Seq数据集,计算各个方法的AUC值需要大量基准基因(即已知差异表达的基因),基准基因的差异表达值需要通过实时荧光是量聚合酶链反应技术(quantitative real-time PCR,qRT-PCR)生物实验来确定.而现实中包含PCR数据的RNA-Seq数据集是极其少的.因此,RobustDEA方法采用了一种相互交叉差异基因集的验证方式来计算各个方法的AUC值,从而避开对PCR数据的依赖.针对每个方法,先计算并排序每个基因的P值,选取前N个基因(N通常选择2 000),这部分基因被认为最有可能的差异表达基因.再计算多个方法的差异基因的交集.交集中基因是被共同认可的差异基因,以此作为该数据集的基准基因,用来计算自身的AUC值,其算法的具体过程如算法1.

算法1:相互交叉差异基因集的验证AUC值计算方法输入:m个方法对差异基因列表(根据P值排序)Repeat:对于方法j 1. 分别选取其它方法的前N个差异表达基因; 2. 前N个差异表达基因的交集; 3. 以交集中基因作为基准基因; 4. 计算AUCj值Until:所有方法的AUC值计算完毕输出:根据各个方法的AUC值计算权重大小
通过AUC值计算的相互交叉验证,可避免PANDORA方法中需要预先使用模拟数据集去计算各个方法权重的过程,不仅可大幅度的提高权重的计算效率,同时也能保留数据集的特点.
1.3 评估准则
选取了多种评估准则来评估不同方法和验证RobustDEA方法的性能.当RNA-Seq数据集包含PCR验证基因,或者将结合方法共同认可的差异基因作为基准基因,选择F1值来评估方法性能.F1值是对精确率(Precision)和召回率(Recall)的加权调和平均,其定义为:
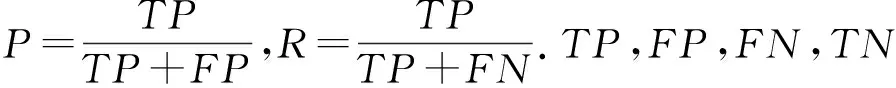
此外,进一步采用ROC曲线和AUC值评估按方法的预测准确性.ROC曲线图是反映横坐标假阳性率(false positive rate, FPR)与纵坐标真阳性率(true positive rate, TPR)之间关系的曲线,其中:
AUC值表示ROC曲线下面积,其值越大表示正样本越有可能被排在负样本之前,即方法的预测性越好.
1.4 RobustDEA实现
为了便于用户使用,RobustDEA方法采用R语言实现,且选择的结合方法都在Bioconductor中提供了R语言包,避免了用户额外安装其它基于Unix的软件,比如Cuffdiff2等.
RobustDEA方法的R语言包和详细文档可以从网址https://github.com/BIO-LAB/RobustDEA下载,供全球生物学家免费使用.
2 实验结果与讨论
2.1 数据集
为了评估RobustDEA方法,文中选择了ReCount数据库中的3个真实RNA-Seq数据集[19],分别是人类大脑数据集MAQC,老鼠数据集Katz和小鼠数据集Hammer.
人类大脑MAQC数据集来自美国药品监督局(FDA)联合全球多所研究机构进行的“生物芯片质量控制”项目[20].MAQC数据集包含2个条件,正常大脑组织(HBR)和病变大脑组织(UHR),每个条件下包含7个重复试验样本,总计包含52 580个基因.过滤掉在所有样本中读段数目都是零的基因,获得11 907个具有表达的基因.此外,MAQC项目提供了1 000个qRT-PCR验证基因.通过筛选具有显著变化的基因(FC<=0.5或FC>=2.0),获得305个验证基因作为标准基因,用来评估不同方法寻找差异表达基因的准确性.
大鼠数据集Katz包含两个实验条件,正常成肌细胞和敲掉CUGBP1的成肌细胞,每个条件包含2个生物性重复实验,总计包含36 536个基因.过滤掉在所有样本中读段数目都是零的基因,获得9 501个具有表达的基因.
小鼠数据集Hammer是关于小鼠脊柱神经发育的研究,包含胚胎发育后2周和2月两个时间点.每个时间点包含2个条件,正常脊柱神经细胞和进行脊柱神经结扎的神经细胞.每个条件包含2个生物性重复样本,总计包含29 516个基因,过滤掉在不同时间点时样本中读段数目都是零的基因,两个时间点分别获得17 125和18 463个具有表达的基因.
2.2 结合方法的选择
目前,提出了大量寻找差异基因表达方法,结合方法的选择会对RobustDEA方法的稳定性和性能影响较大.因此,在不同数据集中性能稳定且时间效率高的方法作为RobustDEA的结合方法的最有选择.根据多个综述性评估不同方法的精确度和时间效率(如表2),文中选择DESeq2(v1.26.0) ,edgeR(v3.28.1),NOISeq(v2.30.0) 和Voom(v3.42.2)4种方法作为RobustDEA的原始方法.PANDORA方法来自R包metaseqR(v1.26.0).
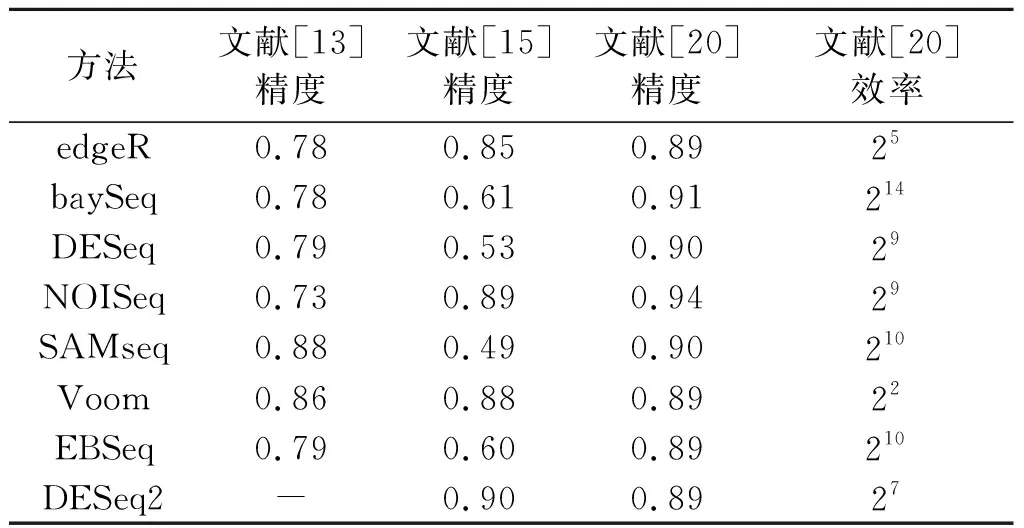
表2 寻找差异表达基因方法在不同数据集上的性能
2.3 MAQC数据集上qRT-PCR标准基因验证
经过qRT-PCR生物实验验证过的基因往往被当做标准基因,用来评估生物信息学中各种方法的准确度.通过注释文件和倍数比率的筛选,MAQC数据集获得了305个qRT-PCR基因,被用来评估不同寻找差异方法的准确性.图2显示不同方法在MAQC数据集上的ROC曲线.其中图2(a)为RobustDEA方法和其他4个原始方法的比较.结果表明,RobustDEA方法和Voom方法的ROC曲线接近重合,但是要明显要好于DESeq2,edgeR和NOISeq 3个方法.这表明在RobustDEA方法在结合不同方法的结果之后,至少可获得不低于原始方法中最优性能.图2(b)为RobustDEA方法与其他3种结合方法的比较结果.从图中可以看出,RobustDEA方法的ROC曲线要明显高于其他3个结合方法.Fisher,Stouffer和PANDORA方法得到比原始方法更差的结果,说明这些方法不够稳定,容易受到原始方法中较差结果的影响.
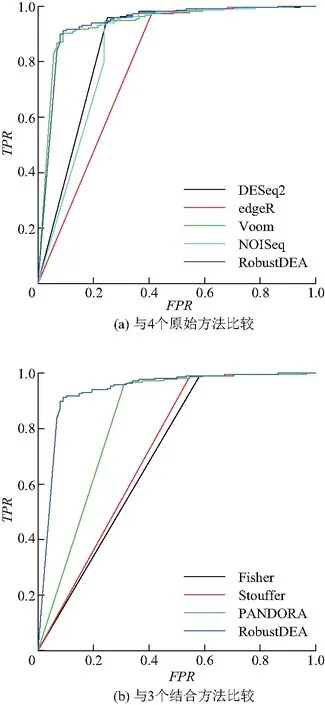
图2 不同方法在MAQC数据集上的ROC曲线
图3为原始方法和结合方法在MAQC数据集上的AUC值和F1值,从这两个值均可说明RobustDEA方法获得了最好准确度,表明该方法相比于其他结合方法,不易受到较差性能的原始方法影响,具有更好的鲁棒性.
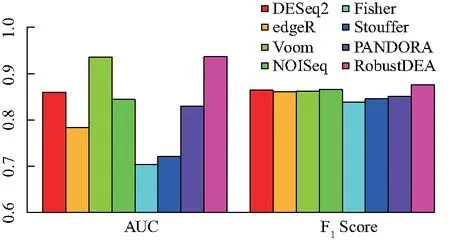
图3 不同方法在MAQC数据集上的AUC值和F1值
2.4 Katz和Hammer数据集的大规模基因验证
MAQC数据集的仅提供了305个qRT-PCR验证基因,缺少大量基因的评估.但是由于qRT-PCR实验的费用问题,现实中缺少包含大量qRT-PCR验证基因的数据集.为了进一步在大规模基因上评估RobustDEA方法的性能,采用交互验证方式构建大规模差异基因数据集[11].首先分别选择4个原始方法各自认为最可能是差异表达的前2 000个基因.将这些基因合并成一个验证基因集,其中4个方法共同认定为差异表达基因作为“真实“差异表达基因,其余为非差异表达基因.
在Katz数据集中,通过交互验证方式构建的验证基因集包含2 501个基因,其中1 517个基因被共同认定为差异表达基因,其余基因即被划分为非差异基因.通过此验证基因集的评估,不同方法的ROC曲线如图4.
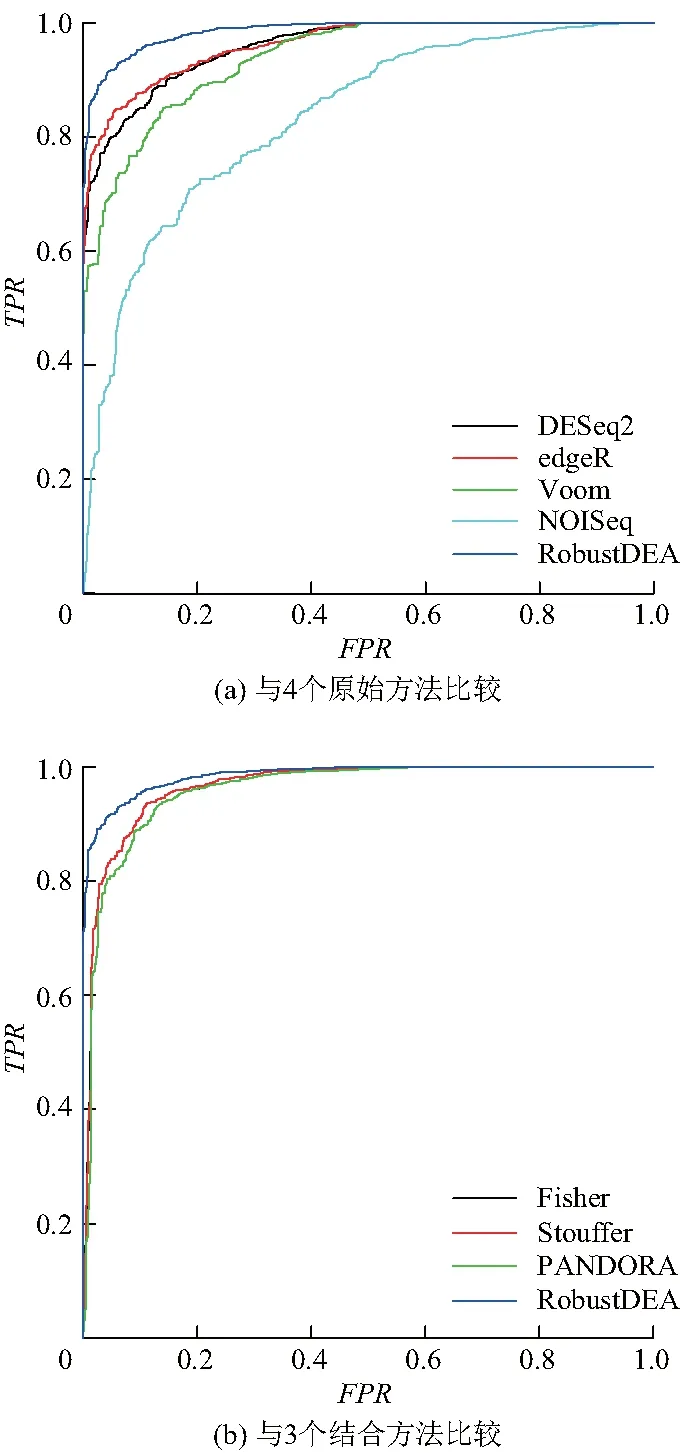
图4 不同方法在Katz数据集中的ROC曲线
从图4(a)中可以看出,RobustDEA方法比4个单独原始方法的性能都要好.从图4(b)可以获得4个结合方法都获得较高的准确度(图4(b)中,PANDORA和Fisher的ROC曲线高度重合).而相比3个结合方法,RobustDEA方法同样表现出最好的性能.
Hammer数据集包含两周后和两月后两个时间点,通过交互验证方式分别构建验证基因集.两周后时间点的验证基因集包含2 540个基因,其中1 484个基因被共同认定为差异表达基因.两月后时间点的验证基因集包含2 995个基因,其中1 088个基因被认定为差异表达基因.在验证基因集中其余基因为非差异表达基因.通过两个验证基因集的评估,图5(a)和(c)显示RobustDEA方法的ROC曲线要高于其他4个单独原始方法,表明RobustDEA方法通过结合不同方法能进一步提升性能.图5(b)和(d)中与其他3个结合方法进行比较,可以看出RobustDEA和PANDORA方法表现稳定,而Fisher和Stouffer方法性能有大幅度下降.
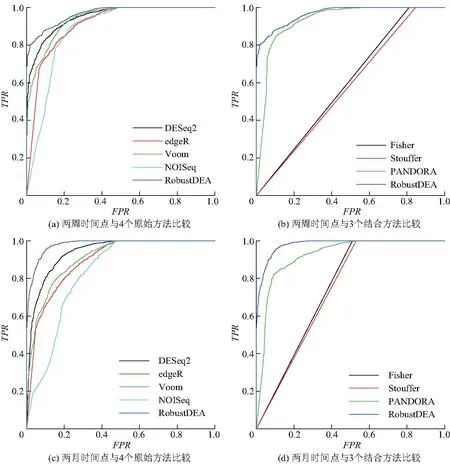
图5 不同方法在Hammer数据集中2个时间点的ROC曲线
表3为不同方法在Katz和Hammer数据集上AUC值,结果表明在3个大规模基因数据集上,RobustDEA方法均可获得最高的性能.虽然PANDORA方法在不同数据集中会比其中某个单独原始方法性能要略低,但是在整体上能获得较为稳定的结果,不会受到性能较差的NOISeq方法的影响.Fisher和Stouffer方法表现极为不稳定,在Katz数据集中能获得极高的准确度,但是在Hammer数据集的2个时间点上性能都大幅度下降.导致此问题的原因在于Fisher方法对不同原始方法赋予相同的权重,容易受到性能较差方法的干扰.而RobustDEA和PANDORA方法对根据原始方法的性能进行加权,可以降低性能较差方法的影响,从而使性能更加表现更加稳定,具有较高的鲁棒性.
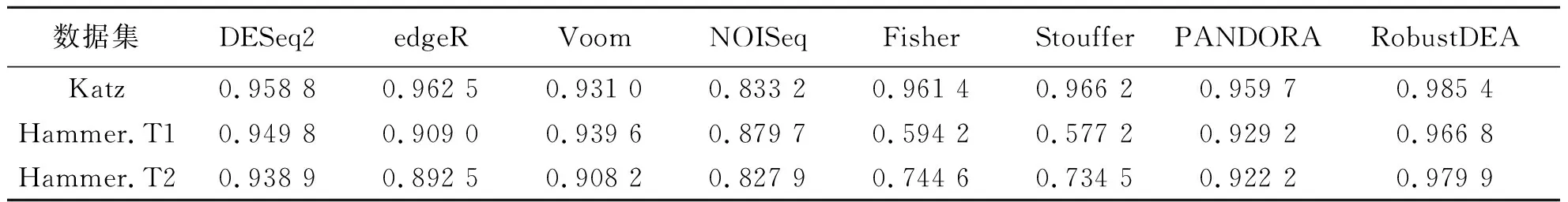
表3 不同方法在Katz和Hammer数据集上AUC值
2.5 时间效率的分析
结合方法的评估过程分为原始方法计算,权重计算和结合评估3个部分.对于所有结合方法来说,原始方法计算所消耗时间都是一致,区别主要在于权重计算部分.PANDORA方法的权重估计是根据现有数据特点去生成模拟数据集,然后再通过计算不同原始方法在模拟数据集上的精度作为权重.这部分的计算需要花费大量时间,导致PANDORA方法的时间效率大幅度下降.并且PANDORA方法的时间消耗与原始方法个数,模拟数据集中样本量,基因个数等参因数有直接影响.RobustDEA方法的权重估计是基于原始方法计算的结果,因此能快速获得不同方法的权重值,极大了提高了计算效率.表4为4个结合方法在MAQC数据集上的计算效率.PANDORA方法设置模拟数据集为4个测序样本,1 000个基因.RobustDEA方法选择N为1 000,其时间为同一工作站的CPU工作时间,性能为2.9 GHz双核和8 G内存,采用单核计算.从表中结果可以看出PANDORA方法花费了整个时间894.6 s中的95%时间进行权重计算,而RobustDEA方法只需要花费5.5 s的时间估计权重值,大幅度提高了效率.虽然RobustDEA方法计算效率仅略高于Fisher和Stouffer方法,但其计算精度和稳定性要明显高于这两个方法.
表4 不同方法在MAQC数据集上计算效率
3 结论
针对不同寻找差异表达基因方法检测得到的差异基因集不一致问题,提出了一种快速鲁棒的RNA-Seq数据寻找差异表达基因的结合方法.通过人类大脑MAQC数据集和多个老鼠数据集的评估,得出如下结论:
(1) RobustDEA方法结合现有寻找差异表达基因方法,通过全新的权重评估方式,可快速获得与当前分析数据集最合适的权重值.
(2) 相比于单个差异表达基因方法和现有结合方法,RobustDEA方法能获得更加准确和稳定的结果.这表明该方法具有较好的鲁棒性,受到性能较差的原始方式的影响较小.
(3) 相比PANDOR方法,RobustDEA方法采用全新的权重估计方式,可大幅度提高计算效率.
今后工作将RobustDEA方法实现为一个可视化在线平台,用户只需导入基因读段数据,平台可快速计算出差异基因集,并提供相应的分析报告,以及更加直观的图表展示,从而让用户使用更加便利.
猜你喜欢
当代陕西(2020年17期)2020-10-28
数学年刊A辑(中文版)(2020年2期)2020-07-25
心电与循环(2020年1期)2020-02-27
数学物理学报(2019年6期)2020-01-13
井冈山大学学报(自然科学版)(2019年4期)2019-09-09
人大建设(2018年5期)2018-08-16
江苏农业科学(2017年5期)2017-04-15
应用科技(2015年5期)2015-12-09
湖北农业科学(2014年3期)2014-07-21
新课程学习·中(2013年3期)2013-06-14
