
化学链合成氨技术研究进展及展望
2021-04-08 12:15冯鸣谦应遥瑶
洁净煤技术 2021年2期
吴 烨,冯鸣谦,方 婧,刘 冬,张 睿,应遥瑶,徐 磊
(南京理工大学 能源与动力工程学院,江苏 南京 210094)
0 引 言
近年来,随着化石燃料的大量消耗和CO2等温室气体的大量排放,氨的无碳燃料属性越来越引起研究人员的重视[1-2]。低能耗高效率的合成氨技术的开发是实现氨燃料化利用的基础。
20世纪初提出的Haber-Bosch法仍是当下工业合成氨的工艺基础,但该方法反应条件苛刻,高温高压的反应工况导致热力学和动力学矛盾突出,因此探索新型“绿色”的合成氨技术一直是科研人员追求的目标[3]。
科技的进步为人们深入探索合成氨机理以及反应物和催化剂的物化性质提供了技术支持。Hoffman等[4]研究了植物固氮酶的固氮机理,发现固氮酶中的关键组分是Fe、Mo或V形成的硫簇。N2加氢生成的N2Hx可能是催化产NH4+过程的中间产物。研究发现,过低的黏附系数是N2在过渡金属表面解离速度慢的主要原因[5-7],提高催化剂表面对N2的黏附系数是促进N2活化的有效途径。Vojvodic和Norskov等[8]理论研究结果合理地解释了过渡金属合成氨的“火山型”活性曲线,为合成氨催化剂的优化改善提供了思路。由光、电等外场力驱动的催化合成氨技术,从降低能耗的角度拓宽了合成氨工艺路线[9-10]。近年来,随着化学链技术的发展,将合成氨分解为吸氮和释氮2个或多个分步反应的化学链合成氨技术逐渐成为研究重点[11]。化学链合成氨过程中,各分步反应可以在不同反应器和反应工况下进行,能够逐一优化使整个反应体系达到最佳效果,有效缓解了反应热力学和动力学之间的矛盾。且反应物交替进料的方式规避了催化合成氨中2个反应物(N2和H2)在催化剂表面竞争吸附问题。氨经济是未来全球能源发展的重要方向,氨在全球氮循环中的作用将越来越重要。
本文概述了合成氨工艺发展历程,总结了近年来多相催化合成氨,光、电等外场力驱动的催化合成氨的研究进展,重点介绍了化学链合成氨的研究现状和潜在的发展方向,以期为清洁高效的合成氨工艺提供参考。
1 Haber-Bosch 式合成氨工艺
1.1 传统Haber-Bosch法合成氨
自1913年,世界上第1座合成氨装置投产以来,现代工业生产中,仍采用Haber-Bosch工艺生产氨(图1)。即在高温(300~500 ℃)和高压(20.26~30.39 MPa)环境中,高纯度的N2和H2在铁催化剂上发生反应合成氨,即

(1)
然而,Haber-Bosch工艺自身的缺点也比较明显,如工作压力高、耗高、转化率低(10%~15%),且CO2排放量占世界总排放量的1%左右[12]。
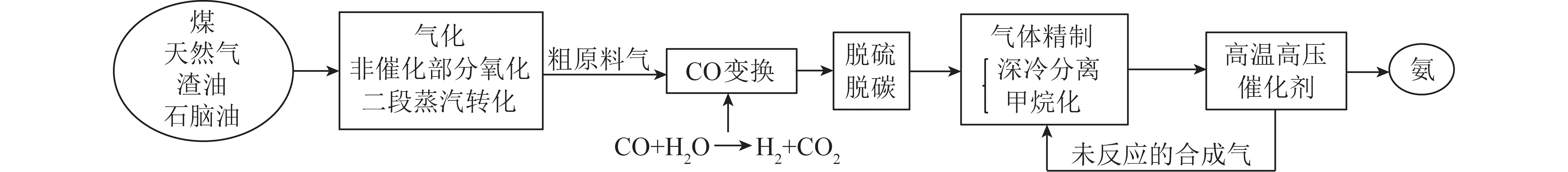
图1 传统合成氨的工艺流程
随着能源与环境压力日益加重,开发更环保的可持续性Haber-Bosch合成氨替代工艺意义重大。以可再生能源为能量来源的低碳排放的合成氨工艺是研究重点[13]。目前,由可再生能源驱动,并通过多相催化、电催化、光催化和化学链等方式进行的“绿色”合成氨路线大致有2条[14]:一是利用可再生能源电解水制得氢气,电解空气制得氮气,氢气与氮气进行反应生成氨(图2中路线1);另一途径则是直接让氮气与水反应生成氨(图2中路线2),该路线可跳过制氢过程,减少中间环节,但目前效率还较低,离实际应用还有较长一段距离。
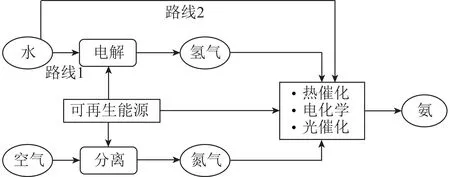
图2 可再生能源驱动的“绿色”合成氨过程
1.2 多相催化合成氨
自Haber-Bosch法合成氨工艺成型以来,研究人员对大多数元素的多相催化合成氨活性进行了深入研究。合成氨反应中过渡金属上的线性关系及火山型曲线如图3所示(EN为过渡金属表面N的吸附能,EN-N为N2解离吸附能垒,ENH、ENH2为结合键能,k为速率常数)。由图3(c)可知,过渡金属的合成氨活性表现为经典的“火山型曲线”,其中位于火山型曲线顶端的VIII族元素Fe和Ru等因适中的物种吸附和解吸能而具有较好的催化合成氨活性,目前工业应用以含多种助剂的熔铁催化剂为主。

图3 合成氨反应中过渡金属上的线性关系及火山型曲线[8,15]
传统Fe基催化剂具有许多优点,如高活性、长寿命和相对较低的成本,但其整体活性受到高NH3浓度下反应率下降的强烈影响,且需要的操作温度及压力较高[16]。而钌基催化剂的主要特点是高活性、高氨浓度以及宽的H2/N2范围[17],并可在低温和较低压力下操作,对水、一氧化碳等杂质不敏感,因此钌基催化剂被称为第2代氨合成催化剂[18]。但由于催化剂成本极高,远超过熔铁催化剂,因此,有待进一步深入研究。非Fe、Ru基催化剂方面研究较多的金属元素是Co和Mo。
由图3(c)可知,Co在火山型的右侧,说明Co的氮吸附能较低,而氮解吸能较高,不是合适的合成氨催化剂。研究人员探究了负载金属氧化物、金属氢化物等方法,来提高Co的合成氨活性。Hagen等[19]研究表明,负载在碳上的钡是提高Co合成氨活性的良好助剂,但仍需高温高压的反应条件才能发挥出较好的催化效果。Gao等[20]研究了BaH2与Co协同催化合成氨的作用(图4),BaH2-Co/碳纳米管(CNT)催化剂在150 ℃以上开始表现出合成氨活性,在300 ℃下,该复合催化剂的活性比BaO-Co/CNT高2个数量级。Yasunori等[21]将12CaO·7Al2O3作为Co催化剂载体,结果表明,载体2.4 eV的低电势能大大降低了Co催化剂合成氨的起始温度。认为低功函数的电极材料给电子可以直接提高Co催化剂的活性。Kojima等发现[22],Co3Mo3N催化剂在相同条件(5 MPa,400 ℃,N2∶H2=1∶3)下的合成氨催化活性比Fe催化剂高。Moszyński等[23]研究了添加铬和钾对于钴钼氮化物催化剂活性的影响,这2种金属添加物的存在使催化剂整体比表面积几乎增加了1倍,当铬质量分数约为1%时,催化活性最大。
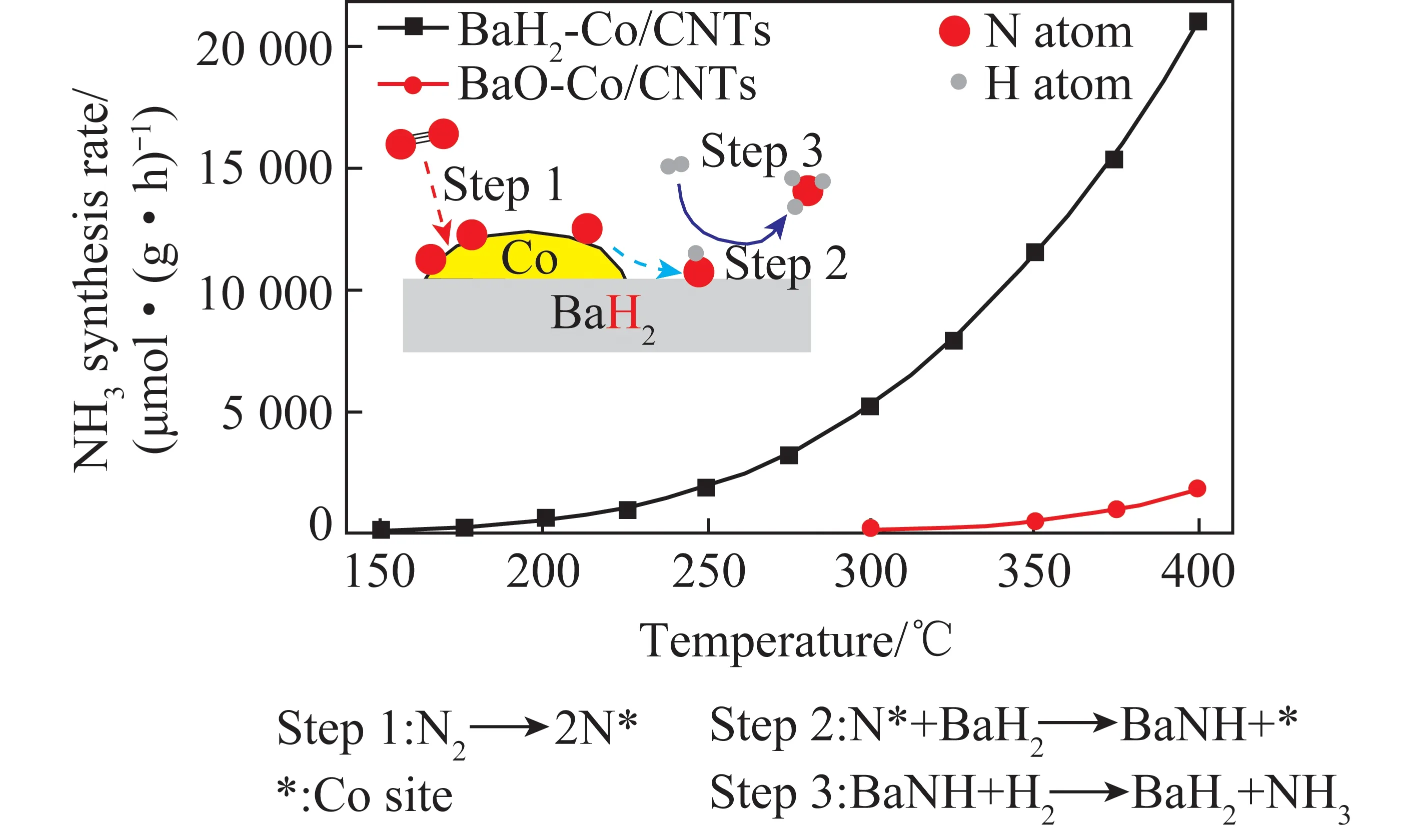
图4 BaH2-Co/CNT和BaO-Co/CNT随温度变化的合成氨速率对比[20]
研究发现,活性金属的结构、金属与载体的相互作用等都能对合成氨反应产生影响。合成氨转化频率(Turnover frequency,TOF)与金属的颗粒尺寸呈正相关,是典型的结构敏感反应[24]。过渡金属上能够活化N2的位点由多核金属原子组成,如相邻的7个Fe原子可能是Fe基催化剂上的活性位(C7位)[25],而5个位于台阶处的相邻Ru原子组成了Ru基催化剂的活性位(B5位)[5]。
在载体方面,碳材料、氧化物、氮化硼、无机电子化物、氧氢化物等新型载体被陆续开发。目前工业用Ru基合成氨催化剂采用的载体是经过高温预处理的一种石墨化碳材料,稳定性很好。Kowalczyk等[26]比较了含石墨的碳和普通活性炭2种钌基催化剂载体,发现含石墨碳的催化剂活性明显优于普通的活性炭。然而,对于碳材料作为载体的合成氨催化剂而言,制约其工业应用的难题是反应条件下载体的甲烷化反应[27],氧化物做载体则可避免这一问题。碱土金属氧化物、稀土氧化物、复合氧化物等是目前主要的研究对象。在400 ℃、0.08 MPa反应条件下,不同载体上Ru催化剂表现出的合成氨性能顺序为:MgO>CaO>Al2O3>Nb2O5≈TiO2[28],其中MgO被认为是有效的合成氨及氨分解反应的载体而得到较多关注。
此外,碱金属、碱土金属氧化物或氢氧化物等物质作为促进剂添加到反应体系中,亦可使活性金属的催化活性得到大幅提高,当碱(土)金属氧化物或氢氧化物的电负性越小时,催化剂的活性越高。碱金属单质具有很强的给电子能力,是钌基催化剂优良的促进剂,其排序为Cs>Rb>K>Na>Li[29]。不同研究人员对于碱土金属的促进机制有着不同解释。Ertl等[30]认为碱金属K是良好的给电子体,可以将电子提供给催化剂以促进N2的解离吸附。而Strongin 等[31]则认为K的存在主要促进了NH3的脱附。
现如今,Fe、Ru等活性金属元素仍然是工业合成氨主要的催化剂。虽然Ru基催化剂的活性高于Fe基催化剂,但其使用成本相应增加。因此,要降低成本,一方面要降低Ru金属的负载量,另一方面要提高催化剂的稳定性及活性。此外,开发新型催化剂以避开过渡金属上吸附物种间线性关系,也是实现温和条件下氨合成的途径之一[32]。
1.3 电化学合成氨
在相对低温、常压环境下,N2(或空气)和H2(或水)在电能驱动下进行的合成氨过程被称为电化学合成氨。该方法可以突破热催化合成氨在热力学上的限制,具有低工作压力、低能耗、清洁无污染等优点,且该过程无CO2排放,对环境较为友好[33]。1985年,Pickett 和Talarmin[34]首次以电化学方法完成了合成氨循环,虽然氨产率和电流效率很低,但实现了常压下合成氨反应,开辟了合成氨研究的新领域。
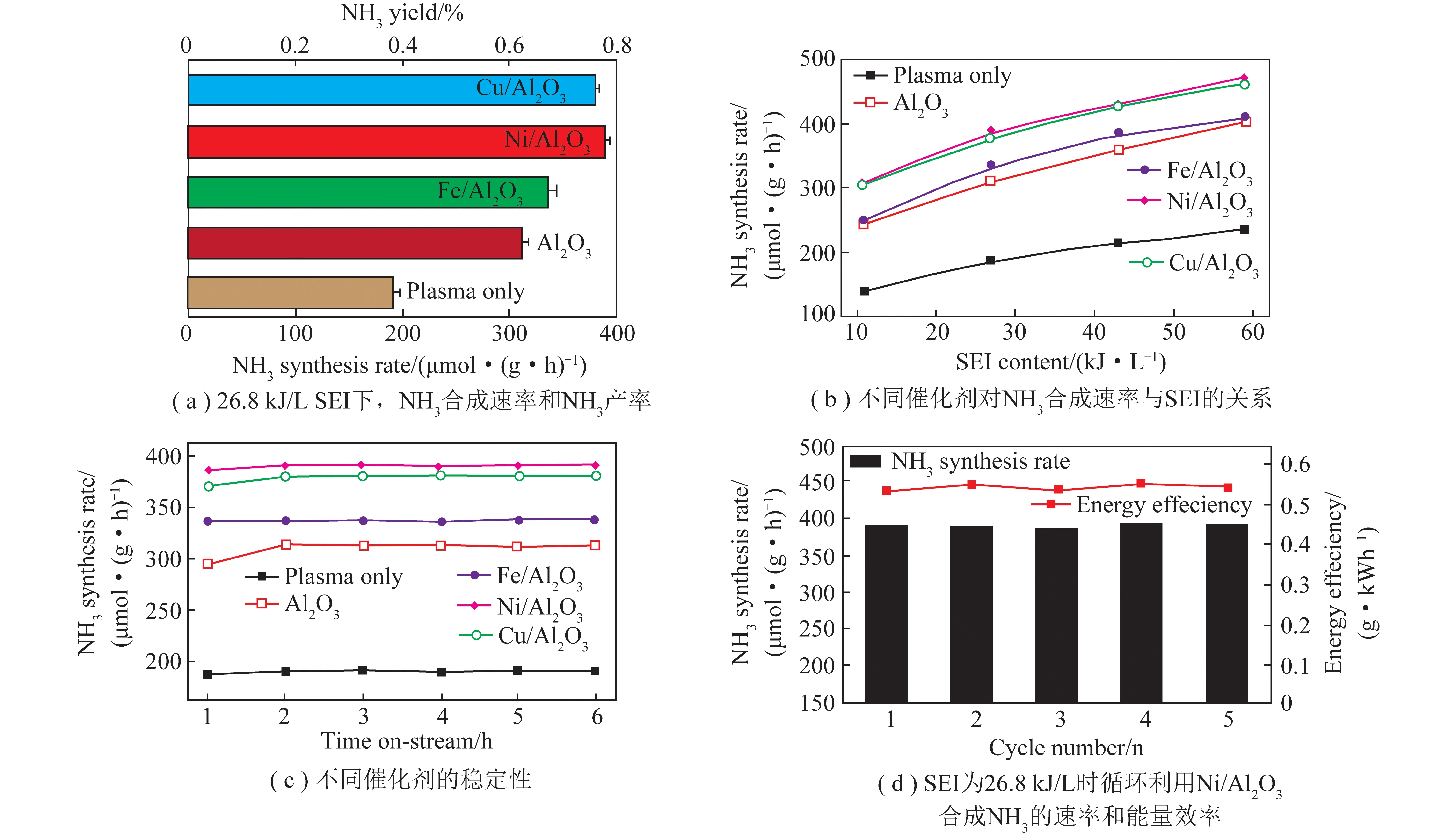
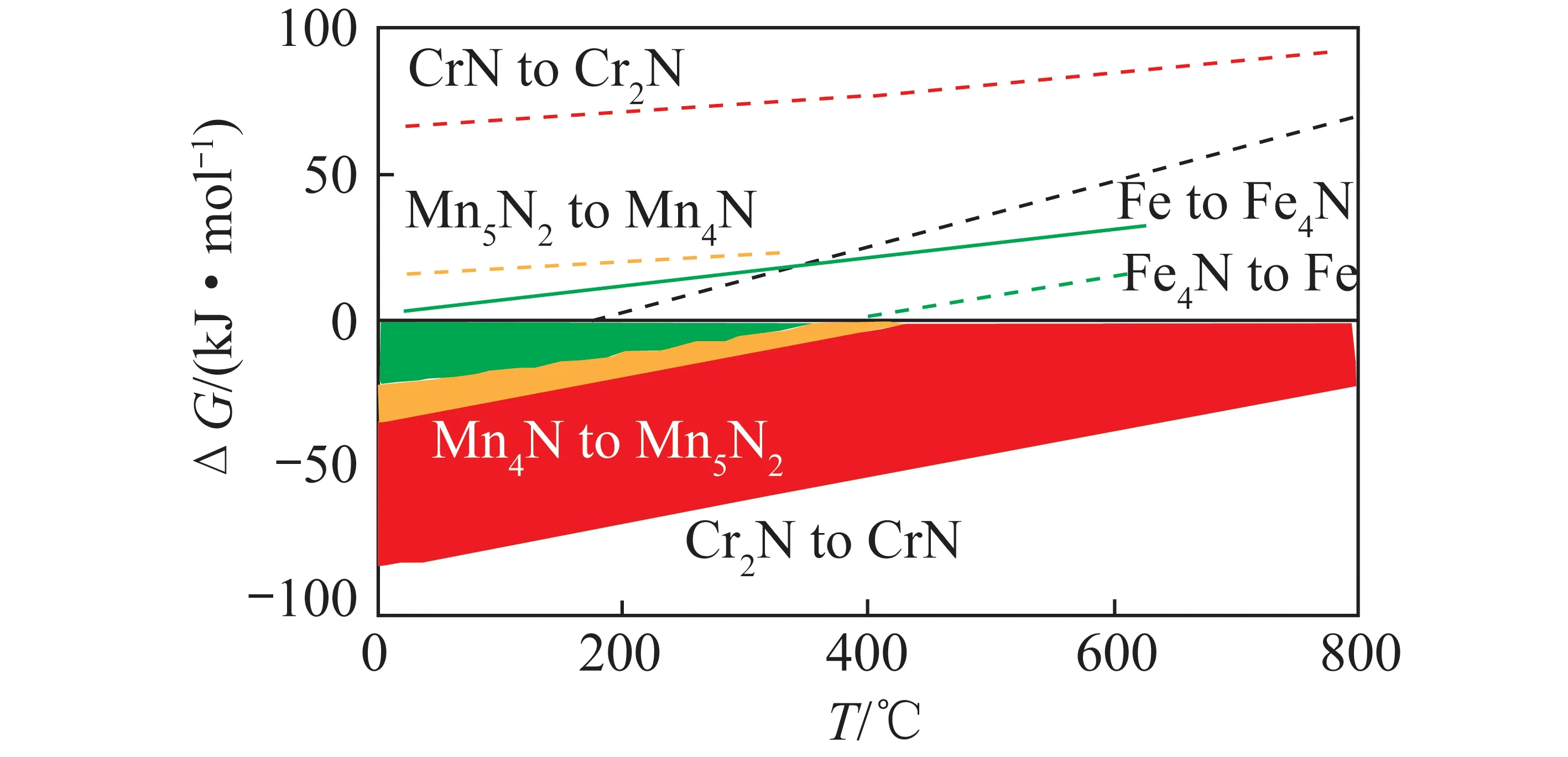
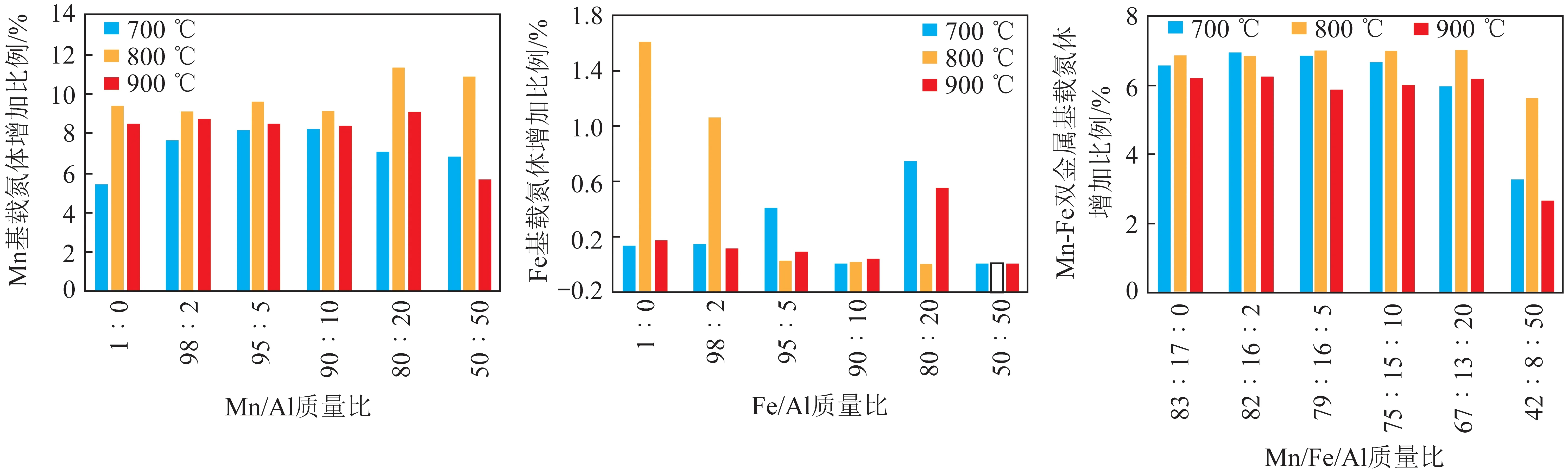
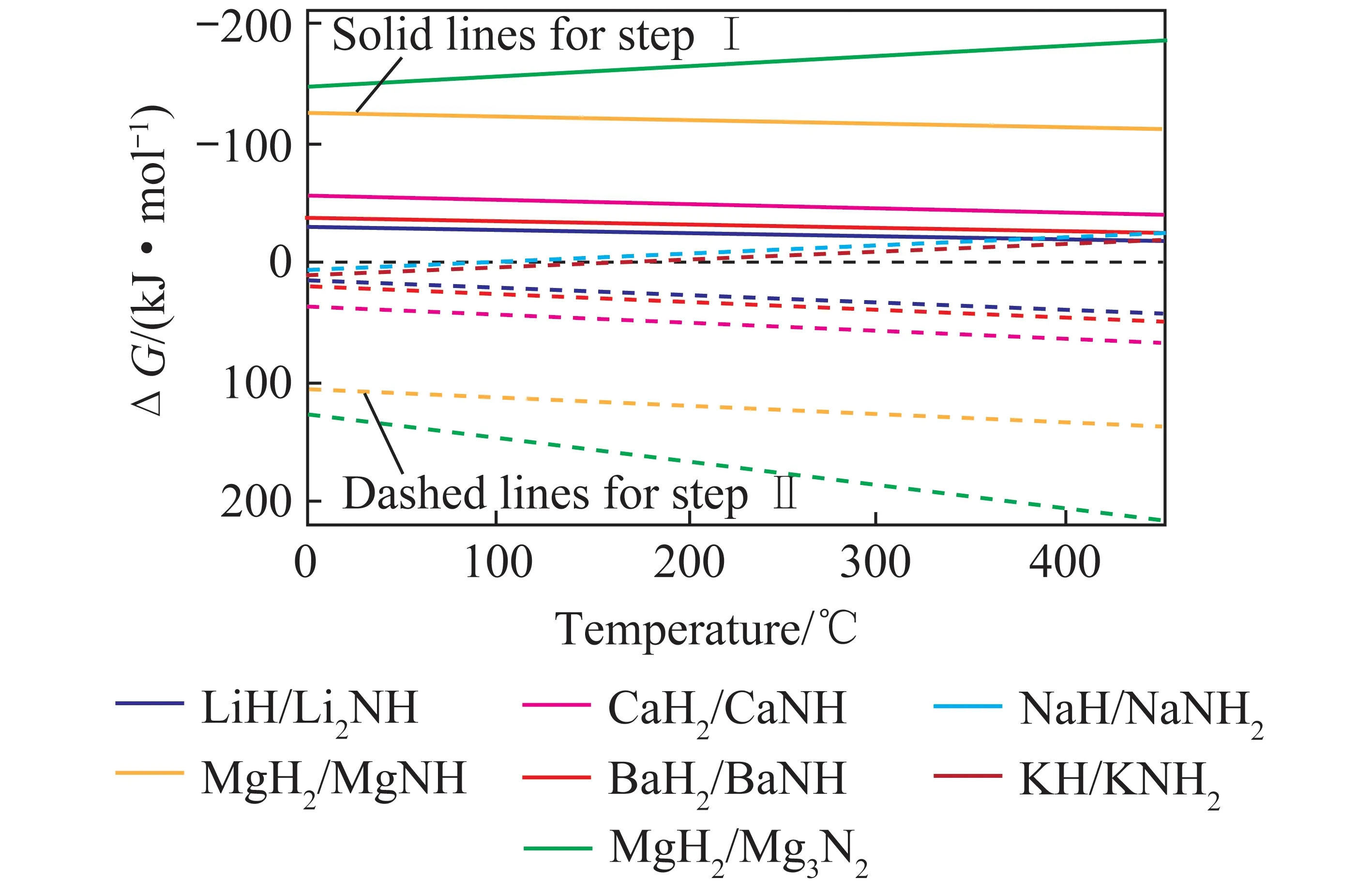
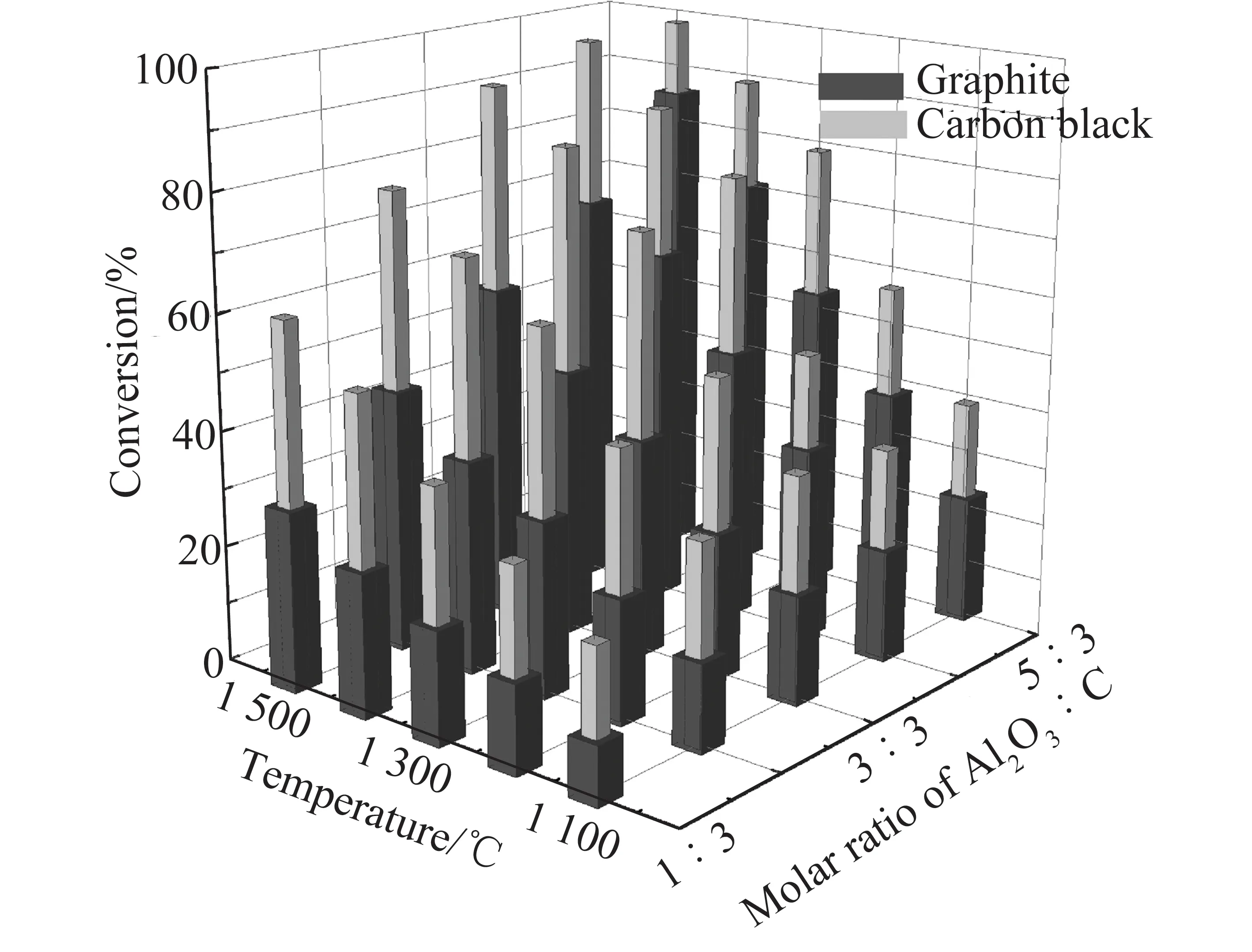
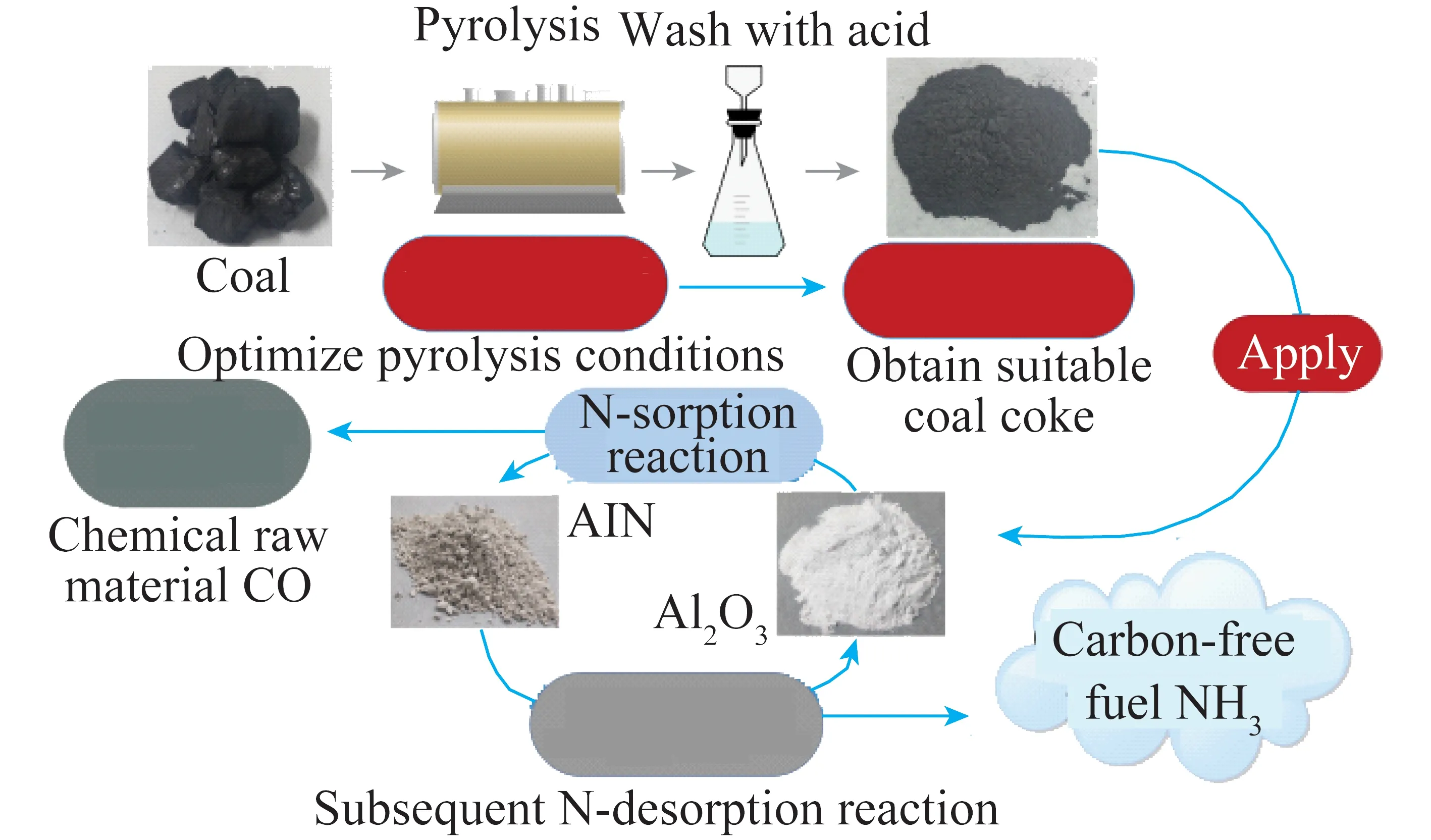
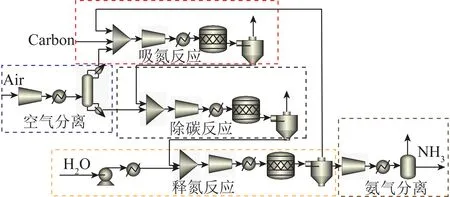
根据电化学合成氨工作温度可将反应分为3类:高温电化学合成氨(t>500℃) 、中温电化学合成氨(100 ℃ 高温电化学合成氨是一种在高温下,以质子导体为电解质的合成氨方法。因其在含氢的高温条件下,表现出良好的质子传导性[35],钙钛矿型陶瓷逐渐引起重视。Marnellos等[36]在1998 年首次报道了以H2和N2为原料,以SrCe0.95Yb0.05O3-δ(SCY)陶瓷膜作电解质,Pd薄膜为阴极和阳极,在双室电解池中常压电化学合成氨,反应装置如图5所示。570 ℃下氨产率最高为5×10-11mol/(s·cm2),电流效率接近75%,这一重大成果推动了电化学合成氨的发展。2005 年,Wang等[37]采用萤石结构型复合氧化物为电解质进行电化学合成氨研究。在520 ℃下,La1.95Ca0.05Ce2O7-δ的最高产氨速率为1.3×10-9mol/(s·cm2),La1.95Ca0.05Zr2O7-δ的最高产氨速率为2.0×10-9mol/(s·cm2)。Li等[38]以Ag-Pd合金作为电极,烧结的钙钛矿型复合物为固体电解质进行电化学合成氨,最高产氨速率可达2.16×10-9mol/(s·cm2)。2015年,Vasileiou等[39]以BaCe0.2Zr0.7Y0.1O2.9(BCZY27)为电解质,Rh膜为阳极,Ni-BZCY27 为阴极,在550 ℃试验条件下电化学合成氨的最高产氨速率为2.9×10-9mol/(s·cm2)。 图5 电化学合成氨反应器[36] 中温电化学合成氨条件较为温和,熔盐电解质体系和复合电解质体系在该领域的应用最为广泛。以熔融LiCl-KCl-Li3N 和LiCl-KCl-CsCl-Li3N 为电解质,Murakami等[40]研究了由N2和各种氢源(包括H2、H2O、CH4、H2S)的电化学合成氨反应[41-46]。发现熔融的氯化物对于中温电化学合成氨效果不佳。2015 年,Kim等[47]在LiCl-KCl-CsCl为电解质的体系中研究了Ti、Fe、Co 和Ni 电极的电化学合成氨性能。4种金属的活性顺序表现为Co>Ni>Fe>Ti,认为N2在熔融氯化物系统中的催化活性由电阻率和润湿性决定。2016年,该研究团队又研究了催化剂对于LiCl-KCl-CsCl电解质体系的影响:当加入纳米Fe2O3时,最高产氨速率可达3.0×10-10mol/(s·cm2),电流效率到达0.14%;当催化剂改为CoFe2O4时,最高产氨速率为1.78×10-10mol/(s·cm2),电流效率为0.17%[48]。Licht等[49]以熔融NaOH-KOH为电解质,悬浮的纳米Fe2O3作催化剂,镍片及200目(74 μm)的蒙乃尔筛网为阳极和阴极,当电流密度为2 mA/cm2、电压为1.2 V时,200 ℃下氨产率为2.4×10-9mol/(s·cm2),合成氨电流效率为35%,同时副产品为H2,产H2速率为6.6×10-9mol/(s·cm2),电解池无须任何隔膜,结构简单。 液体电解质的低温电化学合成氨早在20世纪90年代就有相关报道。1989 年,Furuya等[50]分别以负载Pt和金属酞菁的气体扩散电极为阳极和阴极,以Na2SO4水溶液为电解质, 以N2和H2为原料,通过电化学的方法在常温常压下成功合成了氨,但电流效率很低,这是由N2在水中极低的溶解度以及阴极上存在水电解产生H2的竞争反应造成的。与水相比,脂族醇对N2溶解能力较强,还可以有效抑制水电解产H2反应。Tsuneto等[51]以加入乙醇和LiClO4的四氢呋喃溶液为电解质,在室温下考察了Ti、Mo、Fe、Co、Ni、Cu、Ag 等几种金属阴极的电化学合成氨情况,常压产氨电流效率为5%~8%。当Fe作阴极时,将压力提高至5.1 MPa,电流效率将随之提高至58%,最大氨产率约为4.0×10-9mol/(s·cm2)。Au纳米粒子因其优异的催化性能[52],使人们重新开始关注液体电解质电化学合成氨。将Au纳米颗粒作为催化剂负载于纳米多孔石墨碳上,并以水溶液为电解质,电化学合成氨产氨速率可达4.6×10-9mol/(s·cm2)[53]。与其他贵金属相比,Au的H2分解反应活性较低,可以达到相对较高的电流效率[54]。稀少的存量和高昂的价格使贵金属的应用受到限制,以廉价金属催化剂取而代之势在必行。采用改变SFCN中Fe、Ni含量的方法来影响阴极的催化性能,Xu等[55]获得了目前电化学合成氨领域最高产氨速率1.13×10-8mol/(s·cm2),电流效率为90.4%。 高温质子电解质电化学合成氨虽然克服了高压的条件,但高温会加速氨气的分解。因此,较高的产氨速率却没有使氨的产量得到提升,但电流效率相对较高。中温电化学合成氨条件温和,但中温区的温度依然会使氨发生热分解。针对较高温度下氨分解的问题,Wu等[56]研究表明在反应中加入ZrO2作为催化剂可以对合成氨过程中产生的NH3形成保护作用,从而抑制NH3的分解,提高NH3产率及转化率。低温下电化学合成氨可以实现氨的绿色合成,然而氮氮三键极高的稳定性和氮气在水溶液中较低的溶解度,使常温常压的合成氨在热力学和动力学上都存在巨大的阻碍。另外,析氢竞争反应也会降低氮气还原反应的选择性和产氨速率。通过提高N2的扩散能力,加强传热传质过程,有望改善低温电化学制氨的缺陷。电化学制氨的问题及解决措施如图6所示。 图6 电化学制氨的问题及解决措施 近年来,光催化氮气还原合成氨技术被认为是一种极具潜力的替代Haber-Bosch工艺生成NH3的方法。与传统的工业Haber-Bosch工艺相比,在常温常压条件下,该技术以氮气和水为原料,经太阳能驱动发生氧化还原反应而合成氨,能够实现太阳能向化学能的转化。作为新一代的合成氨工艺,光催化合成氨具有特殊的技术优势:可在常温常压下进行,低能耗,低成本;利用的能源为可再生的太阳能,取代了Haber-Bosch工艺所需的不可再生的化石燃料,降低CO2气体排放量,对环境较为友好;具有绿色可持续的优点;以水作为氢源,地球上的水资源丰富。相比电催化还原制氨,光催化氮还原制氨只需要提供太阳能、水和氮气就可一步法实现氮气的氨转化,而电催化合成氨则需要先将其他能源转化为电能,然后通过电催化过程才能实现固氮过程,因此,光催化制氨具有更低廉的成本。 1977年,Shrauzer等[57]发现在常温常压下,紫外光可以使TiO2表面发生固氮反应。自此,光催化固氮逐渐引起关注。 光催化合成氨主要包括以下步骤:N2在水溶剂中的溶解扩散,N2在催化剂表面的吸附和活化,光生电子还原N2,以及N2逐步加氢质子化生成氨,其中N2的吸附和活化过程是实现从N2到NH3高效转化的最大挑战。基于半导体材料的光催化氮气还原基本的过程如图7所示,包含以下步骤:光激发过程,光生载流子分离、迁移过程以及表面氧化还原反应[58]。为了提升N2的吸附性能,多种富含氧空位的半导体结构陆续开发。Li等[59]将BiOBr纳米片的(001)晶面暴露出来,使该结构中的Bi2O2晶格富含氧缺陷位,通过原位漫反射傅里叶变换红外(FTIR)光谱检测催化剂表面官能团随时间的变化,证实了在可见光下催化剂中氧空位的存在有利于激活氮气,显著提高氮气还原性能。理论计算结果表明,缺陷位能够将N2的键长从原来的1.078×10-10m拉长至1.133×10-10m,削弱了原有的键能,从而起到活化N2的作用,使常温常压下N2的转化成为可能。而Wang等[60]将BiOBr制备成了直径仅5 mm的纳米管,使材料暴露出更多的氧空位。在可见光照射下,该体系可以实现2.3%的量子转化率,NH3生成速率达到了1.38 mmol/(g·h)。Hirakawa等[61]研究发现TiO2在水中的鼓泡反应可实现N2的固定。TiO2表面的Ti3+通过给电子至氮气,可形成化学吸附,成为氮气活化位点。在紫外光激发下,光生电子被表面氧空位捕获,利于Ti3+活化位点的再生。太阳能到化学能的转化效率达到0.02%,转化效率有效提升,但仍低于自然界的光转化效率。 图7 光催化合成氨原理[58] 目前研究者们对于光催化合成氨的化学反应机理认识越来越深刻,但由于该化学反应的综合产率较低,开发高效的合成氨光催化剂和反应体系非常关键,光催化制氨的实际应用尚不成熟。尽管效率不高,但如将光催化反应与其他方式耦合,可能是提升整体反应效率行之有效的方法,可能成为未来发展的重要方向[50]。 等离子体合成氨也是一种有前途的能够代替热催化合成氨的方法,该过程中产生的高能电子和反应性物种,可显著增强反应动力学,使N2活化等热力学不利的反应可以在环境条件下进行。Mehta等[65]利用密度泛函理论研究了Fe、Co、Ru、Rh、Pd和 Ni等过渡金属在Al2O3上的等离子体催化合成氨效果。发现在热催化反应中与氮结合活性较低的过渡金属,在等离子催化反应中有更高的反应活性。然而,在低温等离子体催化合成氨过程中,对于经济、高效、稳定的催化剂研究仍然较少。为了更好了解等离子体-催化剂的相互作用和反应机理,Wang等[66]报道了一种混合等离子体增强催化工艺,使用过渡金属催化剂(M/Al2O3,M=Fe,Ni,Cu)在室温和接近室温的条件下,通过水电极反应器实现了氨的等离子体增强合成。与没有催化剂的等离子体合成氨相比,负载催化剂后显著提高了NH3的合成速率和能量效率,不同催化剂的活性顺序依次为:Ni > Cu > Fe。其中Ni/Al2O3的最高NH3合成速率为471 μmol/(g·h),比仅有等离子体高出1倍,如图8所示。表明催化剂表面的化学效应所提供的催化作用大于物理效应。将金属催化剂Ni负载于Al2O3上,Ni/Al2O3体系产生的等离子体放电更加均匀,既增强了等离子体中N、H和NH的气相自由基反应,也增强了催化剂表面的反应。同时,Ni/Al2O3的表面酸度也发生变化:中酸性位点和强酸性位点减少,这也可能提高了氨的合成速率。近期,Hu等[67]将金属Ru、Co、Ni和Fe与活性碳(AC)结合用于等离子体催化合成氨,结果表明,AC载体上的金属掺杂将NH3的合成效率提高了37.3%。与M/Al2O3体系合成氨促进机理所不同的是,金属掺杂在AC上,使催化剂表面产生了更多的碱性位点,特别是强碱性位点。这些碱性位点加速了气相和M/AC表面上化学反应性自由基和中间体的吸附、扩散和解离,从而增强了等离子体催化过程中NH3的合成。目前,等离子体催化合成氨效率仍然不高,但该方法能够较灵活地与风能、太阳能等可再生能源相结合,是降低能耗成本的可行之策。 将合成氨反应分解为吸氮和释氮2个或2个以上的分步反应,并通过中间产物将各个分步反应依次串联起来实现物质和能量传递的过程被称为化学链合成氨过程。 图8 氨合成催化剂活性的评估[66] 图9 化学链合成氨示意 以传统的Haber-Bosch法合成氨为总包反应,通过载氮体固氮和加氢释氮2个分步反应实现氨合成的化学链合成氨方法,本文称之为Haber-Bosch式化学链合成氨,实现该方法通常为金属氮化物或亚氨基化合物。 2.1.1金属氮化物载氮体 大多数元素都能与N2反应生成氮化物,其中过渡金属氮化物因其具有多种可变价态,能够形成多种类型,且稳定性较差,易于被还原,通常被认为是合适的载氮体材料[68-69]。Michalsky等[70]对部分过渡金属贫氮氮化物继续吸氮形成富氮氮化物的过程和加氢释氮产氨过程进行了试验验证,并结合电子结构理论对上述过程进行了热力学计算。结果如图10所示(G为吉布斯自由能),Cr2N和Mn4N继续固氮生成富氮氮化物CrN和Mn5N2的反应在热力学上易于进行,而富氮氮化物还原为贫氮氮化物的反应热力学阻碍较大;相反,Fe氮化为Fe4N在热力学上较为困难,Fe4N加氢释氮在热力学上可行;氮化锰有多种类型和多样结构,且氮化物之间的转化反应热力学适中,因此大多数研究以氮化锰作为载氮体。 图10 部分过渡金属及其氮化物固氮和加氢释氮反应热力学计算[70] Wang等[71]选择了价格低廉、合成氨性能优良的Fe和有多种价态氮化物的Mn作为载氮体,以H2为氢源,在700~900 ℃成功实现了化学链合成氨,结果如图11所示。结合试验研究和热力学分析分别探究了Fe、Mn单金属基和Mn-Fe双金属基载氮体的氮化和氨化性能,发现Mn的氮化性能好于Fe,当Fe-Mn双金属作为载氮体时,其氮化性能介于2种金属单质之间。在氨化过程阶段,Fe基及Fe-Mn双金属载氮体均能与H2发生反应生成NH3,Mn基载氮体却难以氨化。Fe-Mn双金属载氮体的使用,一定程度上解决了Fe基载氮体氮化困难和Mn基载氮体难以氨化的问题。此外,通过化学反应热力学计算,发现Mn基载氮体的氮化反应是放热反应,而Fe基载氮体的部分氮化反应是吸热反应,2种载氮体混合使用时,放热反应放出的热量可以有效补偿吸热反应的需求,促进了Fe氮化反应的进行,提高Fe基氮化物的产量对于后续载氮体的氨化有积极影响。因此,Fe-Mn双金属载氮体对于整个化学链合成氨有明显的促进作用。 Fig.11 N-sorption capability of N carriers[71] 另外,发现Fe在高温下易烧结,导致N2难以进入金属间隙,阻碍了氮化反应的进一步发生。为缓解这一问题,向载氮体中添加Al2O3作为载体材料,通过增大气固接触面积,优化载氮体的氮化反应性能。结果表明,Al2O3质量分数较小时对氮化反应有积极影响,但随着Al2O3质量分数逐渐增大,由于载氮体占比减小,导致氮化反应性能逐渐下降。表征分析证明,Al2O3的存在使载氮体分散更加均匀,增大了载氮体与N2的接触面积,从而优化载氮体的氮化反应。 虽然以Fe-Mn双金属为载氮体成功实现了化学链合成氨,但该方法仍有很大的发展空间。载氮体氮化和氨化效率仍较低,载氮体的优化值得进一步探索。Al2O3在宏观上促进了载氮体的氮化反应,但效果有限,可以考虑添加对合成氨反应有促进作用的催化剂,从降低化学反应活化能、改善化学反应速率等角度进一步提高反应性能。 2.1.2碱(土)金属氮化物载氮体 2018年,Gao等[72]提出了利用碱(土)金属氢化物与亚氨基化合物之间的相互转化实现固氮及加氢产氨的一项化学链合成氨技术。碱(土)金属氢化物首先进行固氮反应生成相应的亚氨基化合物,随后氢气将亚氨基化合物还原为氢化物,同时释放氨气,具体反应方程式为 (2) (3) 式中,A=Li,Na,K,Mg,Ca,Ba等;x为A的表观价态。 首先通过热力学计算探究各反应进行的难易程度,如图12所示。可知LiH、CaH2、MgH2、BaH2等碱(土)金属氢化物固氮生成相应亚氨基化合物或氮化物的反应在热力学上易于进行, NaH和KH的亚氨基化合物稳定性差,因此难以实现固氮。而亚氨基化合物加氢释氮产氨均表现为热力学不利的反应。综合考虑两步反应的热力学,认为BaH2-BaNH和LiH-Li2NH体系的反应热力学适中,是潜在的能够在温和条件下进行化学链合成氨的材料。 图12 部分碱(土)金属氢化物固氮及相应亚氨基化合物或氮化物加氢产氨反应随温度变化的热力学能[72] 研究发现,LiH和BaH2分别在400 ℃和250 ℃即可进行固氮反应,相应的亚氨基化合物Li2NH、BaNH在300 ℃和200 ℃以上就能实现加氢产氨,结果如图13所示(CL为化学链过程,先通N2再通H2;Catal.为H2、N2同时通入)。这里,氢化物的固氮机理与过渡金属固氮机理显著不同:氢化物中的负氢物种(H-)作为电子供体活化N2并生成H2,而过渡金属本身作为电子供体还原N2。同时发现,Fe、Co和Ni等3d过渡金属对氢化物固氮及亚氨基化合物加氢产氨有显著催化和加速效果。另外,对比了在管式炉中交替通入N2和H2与催化合成氨的产氨速率的高低,发现化学链合成氨产氨速率远高于催化合成氨,且开始产氨的温度节点更低。研究者认为,化学链合成氨交替固氮和加氢释氮的方式极大避免了N2和H2在催化剂表面竞争吸附问题,是其显著优势。 图13 金属亚氨基化合物载氮体[72] 2007年,Gálvez等[73-75]提出了一种以AlN为载氮体的化学链合成氨方法,该方法通过氧化铝的碳热还原反应(吸氮反应)和氮化铝的水解反应(释氮反应),在实现氧化铝和氮化铝相互转化的同时,将碳源的能量转移到NH3中,反应方程式为 (4) (5) 对于这2个分步反应,众多科研工作者从不同角度进行了细致深入的研究。 2.2.1吸氮反应 针对吸氮反应,即氧化铝碳热还原反应,国内外学者从反应工况、反应物种类、催化剂等不同角度进行了深入研究。Forslund等[76]使用石墨炉研究了反应工艺参数对反应过程的影响,发现反应速率随着温度和压力的上升而提高。Tsuge等等[77]提出1 500 ℃下,氧化铝的反应活性以γ-Al2O3>Al(OH)3>α-Al2O3的规律排列,而国内学者匡加才等[78]发现氮化率按Al(OH)3>水铝石>γ-Al2O3的顺序排列。Baik等[79-80]发现,由于蔗糖在高温下分解产生的多孔碳增大了与Al2O3的接触面积,从而促进了Al2O3的氮化。Takayuki等[81]发现,当向反应体系中添加CaF2时,CaF2会与Al2O3反应生成中间物CaO·6Al2O3(CA6)和CaO·2Al2O3(CA2),促进反应的进行。Fu等[82]研究了Y2O3对吸氮反应的影响,发现反应过程中氧化铝会先与Y2O3反应产生铝酸盐(Y-aluminates),再与碳源及N2进行碳热还原反应,将固-固反应转变为固-液反应,极大促进了反应的进行。 Zhang等[83]探究了石墨和碳黑2种不同类型的碳源对于吸氮反应的影响,得到了较为合适的吸氮反应工况。不同温度下碳消耗率与Al2O3∶C摩尔比的关系如图14所示,碳黑在吸氮反应中的反应活性远优于石墨,这是由于碳黑中含有大量无序化结构的碳,而石墨中的碳具有稳定的晶格结构,反应活性较差。发现提高反应温度能促进反应的进行,但过高的温度会破坏反应产物的孔隙结构,导致反应产物的比表面积大大降低,不利于释氮反应的进行。综合考虑,认为反应温度为1 200 ℃,活性氧化铝与碳黑的摩尔比为3∶3,是化学链制氨工艺中吸氮反应较合适的工况。Feng等[84]在此基础上,研究了如何将煤炭高效应用于碳基化学链合成氨。研究发现,预处理后煤焦的无序化程度是影响吸氮反应中碳转化率的主要因素,在升温速率为10 ℃/min,700 ℃的CO2气氛下,热解得到的烟煤焦能较高效参与碳基化学链合成氨中的吸氮反应(图15)。 图14 不同温度下碳消耗率与Al2O3∶C摩尔比的关系[83] 图15 煤炭作为碳源的化学链合成氨[84] 2.2.2释氮反应 AlN通常被用作陶瓷和半导体材料,对于稳定性要求较高,因此众多学者针对AlN的抗水解特性进行了大量研究。李远强等[85]发现含有AlN粉末溶液的pH值先缓慢上升,后迅速上升,最后趋于稳定。推测氮化铝水解产生氨气溶于水后,碱性的氨水又促进了氮化铝的水解。Andraz等[86]测定了氮化铝颗粒在室温及40~90 ℃恒温水浴下溶液pH的变化规律,其趋势与李远强等所得结果基本吻合,这种羟基促进氮化铝水解的现象被称为“碱催化效应”。Bartel等[87]通过密度泛函理论探究了氮化铝水解初始阶段的反应机理。结果表明,Al—N键的断裂是影响氮化铝水解速率的关键步骤,氮化铝水解的主要机理是羟基介导的质子转移。羟基的存在很好地降低了氮化铝水解的活化能,提高了反应动力学,从微观角度证实了氮化铝水解过程中的碱催化现象。 参考前人研究结果,Wu等[88]首先对比了8种常见的催化剂对于AlN水解反应的影响,结果如图16所示(X为增重比例)。发现Fe2O3对于释氮反应具有良好的催化效果。当Fe2O3的负载量为5%时,AlN的起始水解温度能降低100 ℃。阿弗拉米-艾罗费夫动力学模型显示,Fe2O3使释氮反应的活化能由403.9 kJ/mol降至300.2 kJ/mol,反应级数由1.5降低至1,极大促进了AlN的水解。并结合密度泛函理论计算,发现H2O分子极易在Fe2O3表面发生解离吸附,形成表面羟基,从而提高AlN水解的动力学。然而,由于Fe2O3对NH3的分解具有一定的促进作用,导致NH3收率较低。 图16 AlN释氮性能[88] Zhao等[89]在研究TiO2对钾基吸附剂捕集CO2的催化机理时,发现H2O分子极易在TiO2表面吸附解离,形成游离的羟基。另外,Aapera[90]、Henderson等[91]利用密度泛函理论也证实了这点,H2O分子在TiO2表面吸附解离反应为 (6) Gao等[92]将TiO2与AlN混合,利用固定床试验台探究了TiO2对释氮反应的影响。发现金红石型TiO2对于AlN的水解反应有促进作用,虽然催化效果略低于Fe2O3,但TiO2不会促进NH3分解,因此NH3产量明显提高。 由于NH3稳定性差,高温下易分解,导致NH3实际产量远低于理论产量,因此,探索抑制NH3分解的方法对于化学链合成氨工艺十分有益。Wu等[56]将ZrO2作为催化剂与AlN混合,900 ℃反应温度下,当ZrO2添加量由0增加至80%时,AlN反应量没有明显提升,但NH3产率却由40%提高至80%,结果如图17所示。同时,对NH3在ZrO2表面的吸附行为进行了量子化学计算,发现NH3容易在ZrO2(001)表面形成分子吸附,减少了NH3自由分解的路径,显著提高了NH3的产量。 图17 900 ℃下不同ZrO2负载量下NH3产量(nN)、AlN转化量(nA)和相应的NH3产率(η)的变化情况[56] 2.2.3经济性分析 南京理工大学吴烨老师课题组利用Aspen Plus软件,基于试验研究结果,对化学链制氨基本系统进行了建模和优化,如图18所示。流程模型包括吸氮反应、释氮反应、空气分离、除碳、氨气收集5个部分,该流程模型可得到质量分数99.733 6%、压力2.16 MPa的液氨产品。 图18 碳基化学链制氨工艺流程模型 此外,通过对流程模型的能耗分析,得到未优化的模型的能耗为32.2 GJ/t(以NH3计,下同),且分析结果表明,释氮反应模型和除碳模型是潜在的可优化目标。加入换热器模块Heat X将废气携带的热量回收用于反应物料的预热,有效降低了工艺流程的能耗,优化后能耗降为11.7 GJ/t。 2.2.4展望 碳基化学链制氨作为一种新型合成氨方法,较有效解决了催化合成氨热力学和动力学的矛盾。但是针对两步反应的研究大多都相对独立,少有将载氮体循环应用于两步反应,探究载氮体循环吸释氮稳定性和效率等问题的研究。另外,针对两步反应对温度要求均较高的问题,可采用太阳能聚热供能,同时以生物质炭为碳源,从而达到“绿色”合成氨的效果(图19)。 图19 碳基化学链“绿色”合成氨工艺 氨作为可再生清洁能源的潜力逐渐被发掘,“绿色”高效合成氨工艺一直是业界永恒的课题。传统Haber-Bosch式合成氨的热力学和动力学矛盾突出,催化和表面科学以及理论计算的进步,帮助人们更深入了解催化合成氨的反应机理,推动了多相催化、电催化、光催化、等离子体催化等低温合成氨技术的发展。 化学链技术为合成氨技术提供了新的发展方向,化学链合成氨在缓解催化合成氨动力学与热力学矛盾,规避反应物竞争吸附问题上表现出鲜明的特点。Haber-Bosch式化学链合成氨在转化效率方面具有较好的优越性,但以H2为氢源导致工艺成本较高。以金属氧化物为载氮体的碳基化学链合成氨虽然产氨效率相对较低,但氢的来源是H2O,并能将化石燃料中的能量转移至NH3中,副产物CO是重要的化工原料,从而达到了无碳利用化石燃料的目的。载氮体作为化学链合成氨过程的核心,其性能直接影响反应效率。随着先进材料科学的发展,对于化学链合成氨对载氮体要求的了解将更加深入,性能优良的载氮体材料将引领化学链合成氨技术取得新的突破。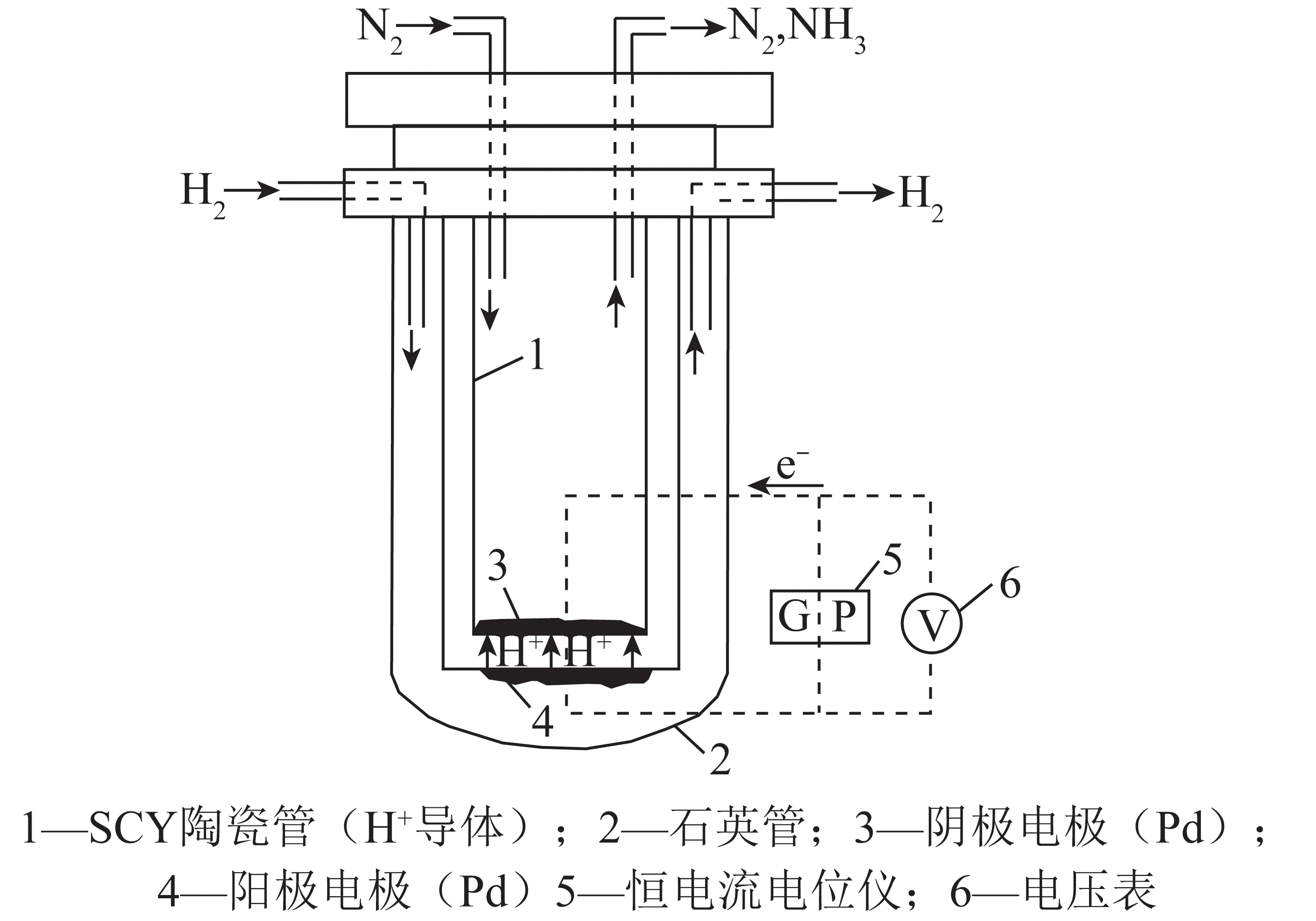
1.4 光催化合成氨


1.5 等离子体催化合成氨
2 化学链合成氨
2.1 Haber-Bosch式化学链合成氨


2.2 碳基化学链合成氨




3 结 语
猜你喜欢
中国化肥信息(2022年4期)2023-01-02
中学生数理化(高中版.高考理化)(2021年4期)2021-07-19
陶瓷学报(2021年1期)2021-04-13
粉末冶金技术(2021年1期)2021-03-29
表面工程与再制造(2019年6期)2019-08-24
热处理技术与装备(2019年1期)2019-03-14
石油化工自动化(2018年5期)2018-11-14
电子制作(2018年12期)2018-08-01
资源节约与环保(2018年1期)2018-02-08
池州学院学报(2017年3期)2017-10-16
