
小反刍兽疫病毒芯片式数字PCR检测方法的建立及应用
2021-12-11 05:18杨鸣发马云云于志亚刘家森康洪涛曲连东
中国动物检疫 2021年12期
杨鸣发,马云云,于志亚,刘家森,康洪涛,姜 骞,曲连东
(1.中国农业科学院哈尔滨兽医研究所,兽医生物技术国家重点实验室/自然疫源性人畜共患病团队,黑龙江哈尔滨 150001;2.东北农业大学动物医学院,黑龙江哈尔滨 150030)
小反刍兽疫(peste des petits,PPR)是由副黏病毒科麻疹病毒属的小反刍兽疫病毒(peste des petits ruminants virus,PPRV)引起的一种急性烈性传染病,在我国和许多国家都有报道,可导致绵羊和山羊出现急性发热、眼鼻分泌物增多、口腔炎症、腹泻和肺炎等症状[1]。PPRV 基因组全长为15 948 nt,为不分节段的单股负链RNA 病毒,其6 种基因编码的8 种蛋白分别为N-P(C/V)-M-F-H-L。其中,N 蛋白为病毒核蛋白,是含量最高的编码蛋白,具有保护病毒RNA 免受核糖核酸酶I 破坏的功能,与RNA 结合作为病毒转录的模板[2]。PPRV于2007 年首次在我国被发现[3]。PPRV 宿主范围广,可感染包括山羊和绵羊在内的多种小反刍动物,野生动物亦可感染,骆驼和水牛也有被PPRV 感染的报道[4]。对于PPR 的诊断方法目前以病毒分离鉴定、中和试验及实时荧光定量PCR(quantitative real-time polymerase chain reaction,RT-qPCR)为主。前两种方法所需时间较长,不适于PPR 的快速诊断,而RT-qPCR 具有敏感性高、特异性强、检测时间短、无须核酸电泳等优点,是传统PCR不能比拟的。相比于RT-qPCR,芯片式数字PCR(chip-based digital polymerase chain reaction,cdPCR)在单分子层面上的检测为绝对定量,可以彻底摆脱对标准曲线的依赖而直接给出靶序列的拷贝数,从而提高了试验结果在批内和批间的稳定性,甚至能用来对标准品进行定标。cdPCR 已经实现了高通量样品条件下的自动化操作,同时可以从大量样品中直接定量核酸,更适合复杂背景中的突变检测和高污染样品中的核酸定量,从而提高核酸检测的准确性和灵敏度[5-6]。为此,本研究以PPRV-N基因的保守区域序列为研究对象,采用cdPCR 技术进行PPRV 检测,并通过优化反应条件及特异性、敏感性、重复性等一系列试验,初步建立了PPRV cdPCR 检测方法。该方法具有高通量、绝对定量、快速、简便、特异性强、重复性好、灵敏度高等优点,从而为PPR 的早期诊断、流行病学调查和检验检疫提供了高效检测方法。
1 材料与方法
1.1 材料
PPRV Nigeria75/1 疫苗株核酸,由中国动物卫生与流行病中心馈赠;犬瘟热病毒(canine distemper virus,CDV)、新城疫病毒(Newcastle disease virus,NDV)、山羊痘 病 毒(goatpox virus,GTPV)、副流感病毒5 型(parainfluenza virus 5,PIV5)和口蹄疫病毒(foot-and-mouth disease virus,FMDV)等核酸,由中国农业科学院哈尔滨兽医研究所(CAAS-HVRI)兽医生物技术国家重点实验室/自然疫源性人畜共患病团队保存。用于检测的绵羊和山羊临床样本47 份,储存时间为2016—2017年,均由本实验室灭活处理保存。
QuantStudio™3D 数字PCR 仪和QuantStudio™3 和5 荧光定量PCR 仪,均购自美国赛默飞公司。QuantStudio™ 3D 数 字PCR Master Mix,产自Thermo Fisher(美国);AxyPrepTM质粒提取试剂盒,产自QIAGEN(德国);SuperScript III/PlatinumTaqOne-step RT-PCR Kit、MLV 反转录酶、MiniBEST Agarose Gel DNA Extraction Kit 胶回收试剂盒,均购自Takara 公司。
1.2 引物和探针
应 用MegAlign 软件对GenBank 中所有的PPRV 全基因组序列进行比对,选取N基因中特异性编码区保守序列设计引物和探针。利用Primer Premier 6 对设计的引物(PPRV-N-F:CTCGGAAATCGCCTCACAGAC;PPRV-N-R:CAAATGGGTCCGAAGGAAGG)进行分析,预期扩增片段大小为166 bp,探针序列为FAMCGCCTACACCAGCGACCAGAGAAGAAG-BHQ(探针5'端标记荧光基团FAM,3'端标记荧光淬灭基团BHQ)。所有引物和探针均由吉林库美生物科技有限公司合成。
1.3 标准品制备与稀释
采用反转录试剂盒添加M-MLV 逆转录酶,以上游引物反转录合成cDNA。对利用PPRV Nigeria 75/1 疫苗毒株设计的N基因引物进行序列扩增,扩增产物预期大小为166 bp;使用TaKaRa MiniBEST Agarose Gel DNA Extraction Kit,将PCR产物连接至pMDTM18-T 载体,得到重组质粒pMDTM18-N。使用分光光度计测定标准质粒浓度。质粒浓度计算公式为
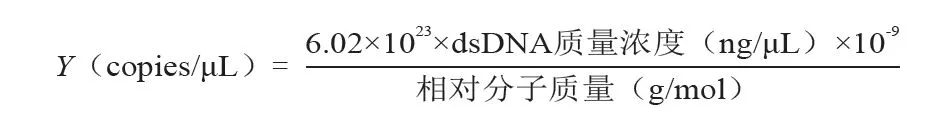
本研究标准质粒浓度为1.22×1012copies/μL,相对分子质量为58 040 g/mol。用ddH2O 将标准质粒进行10 倍系列稀释,共11 个稀释度(1.22×1012~1.22×102copies/μL),以此构建RTqPCR 标准曲线。
1.4 cdPCR 反应体系和条件优化
在QuantStudio™ 3D 数 字PCR 仪上进行cdPCR。配制反应混合液体系:3D Digital PCR Master Mix 7.5 μL,上下游引物各1.0 μL,探针1.0 μL,ddH2O 1.5 μL,cDNA 模 板3.0 μL,最终体 积 为15.0 μL。将固态芯片放在固定器上,吸取配制好的反应液均匀滴涂在芯片上,然后将芯片插入加载器上紫外胶连,插入后15 s,取出芯片。将芯片放入QuantStudio 3D Digital PCR Instrument 仪进行扩增。扩增条件:96 ℃ 10 min;60 ℃ 2 min,98 ℃30 s,39 个循环;60 ℃ 2 min,4 ℃ 60 min。扩增完成后,利用QuantStudio™ 3D AnalysisSuite™软件进行数据分析。每个样本重复试验3 次。利用QuantStudio™ 3 和5 Real time PCR 仪进行RTqPCR。反应程序:50 ℃ 10 min;95 ℃ 2 min;95 ℃ 1 s,60 ℃ 20 s,共40 个循环。反应总体系 为25.0 μL,其中质粒3.0 μL,SuperScript III/PlatinumTaqOne-step RT-PCR reaction mix 12.5 μL,SuperScript III/PlatinumTaqOne-step RT-PCR enzyme mix 0.5 μL,ROX 染料0.5 μL,探针(5 pmol/μL)0.5 μL,上下游引物(10 pmol/μL)各1.0 μL,无核酸酶水6.0 μL。每个样本重复试验3 次。
1.5 敏感性试验
为确定检测阈值,将制备的PPRV cDNA 标准品连续作10 倍稀释(1.22×105~1.22×10-3copies/μL 9 个浓度梯度)进行cdPCR 检测,并将cdPCR 检测方法与RT-qPCR 进行比较,每个稀释度样品重复3 次。
1.6 重复性试验
分别取PPRV cDNA 标准品连续作10 倍稀释(1.22×104~1.22×100copies/μL 5 个浓度梯度)进行cdPCR 和RT-qPCR 检测,分析组间重复性,不同稀释度3 次重复,同时进行组内重复性评价。
1.7 特异性试验
利用建立的cdPCR 方法引物分别检测CDV、NDV、GTPV、FMDV、PIV5 等病毒 的DNA 或cDNA,以PPRV 标准品1.22×105copies/μL 的cDNA 作为阳性对照,ddH2O 作为阴性对照,进行特异性评价。
1.8 临床样本检测
采集2016—2017 年黑龙江省牧羊场47 份临床样本,包括肺、肠、脾、淋巴结和血液。所有临床样本提取组织RNA 反转录成cDNA 进行PPRV RT-qPCR 和cdPCR 检测,记录阳性检出率,比较两种方法的检测灵敏度。
2 结果
2.1 引物筛选及标准品构建
选取PPRVN基因编码区序列同源性和保守性较高的区域设计引物。引物序列覆盖39 株N基因编码区核苷酸(上游引物起始位点PPRVN-F:1182~1202;下游引物起始位点PPRV-N-R:1327~1347),与OIE 推荐验证的引物位点相似(图1)。经2%琼脂糖凝胶电泳分析,RT-qPCR产物显示扩增片段大小为166 bp。将PCR 产物克隆到pMDTM18-T 载体后构建重组质粒PPRV dsDNA 标准品。质粒pMDTM18-N 标准拷贝数为1.22×1012copies/μL。
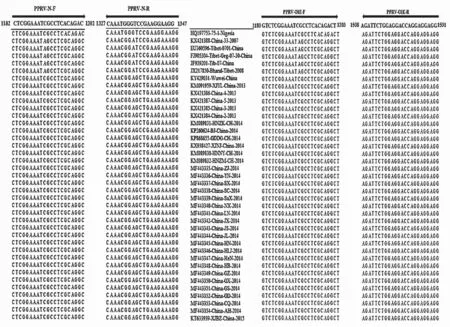
图1 PPRV N 基因引物靶序列的位置
2.2 RT-qPCR 标准曲线建立
将标准质粒pMDTM18-N 连续10 倍稀释构建标准曲线进行RT-qPCR。以质粒拷贝数的对数为横坐标,Ct 值为纵坐标,绘制标准曲线,建立的标准曲线相关系数R2为0.990,斜率为-2.33,截距为37.068(图2)。
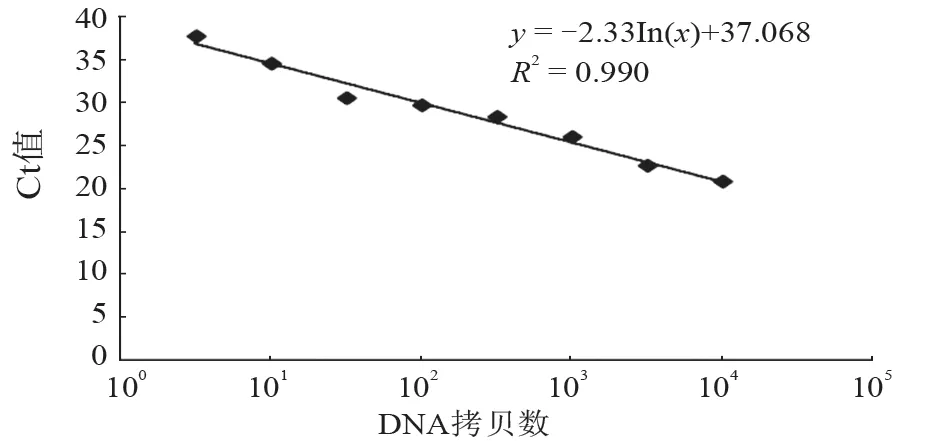
图2 建立的RT-qPCR 标准曲线
2.3 cdPCR 反应条件确定及判定
图3-A 为芯片的扫描结果散点图,蓝色为阳性反应单元,黄色为阴性反应单元。由图可见,反应体系均匀涂布整个芯片,且封闭制作质量较好。散点图(图3-B)用两种颜色表示,分别显示了FAM 荧光基团扩增与未扩增的PCR 孔分布。选取FAM 和BHQ 两种荧光通道,可以在平面上显示两种荧光的分布强度,黄色对应于较低荧光的未扩增孔,而蓝色对应于明显较高荧光的扩增孔。未扩增孔和扩增孔之间的分布具有良好的区分性。统计柱形图(图3-C)显示了芯片阴性反应单元和阳性反应单元的分布,可以看出阴性和阳性反应单元之间形成独立的峰型。使用软件分析1.22×(105~102)copies/μL 4 个稀释度的标准品,结果见图3-D。每个稀释度作3 次重复,在每组中计算平均值的标准误差(SEM),以估计3 个试验的方差,每个样本代表试验独立性(P<0.05)。
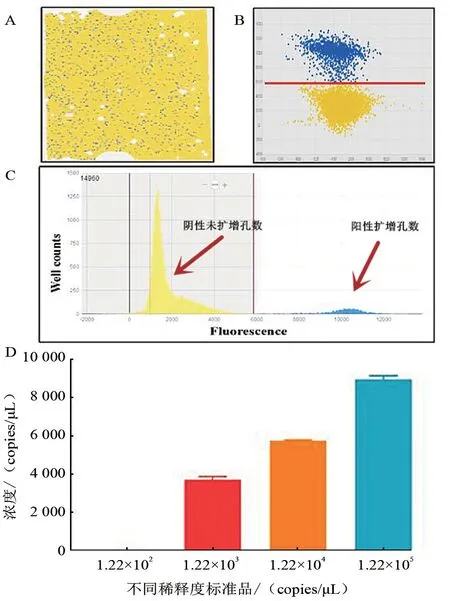
图3 cdPCR 反应条件建立
2.4 敏感性试验
将标准品pMDTM18-N 连续进行10 倍稀释(1.22×1012~1.22×102copies/μL)后进行PCR 检测,发现本试验设计的cdPCR 检测引物最低灵敏度为1.22×102copies/μL(图4-A)。比较OIE 推荐引物的灵敏度1.22×105copies/μL(图4-B),显示cdPCR引物敏感性高于OIE推荐引物1 000倍。cdPCR 在每个15.0 μL 反应体系中,最低灵敏度为(0.44±0.14)copies/μL。RT-qPCR 的检测 限 为1.22×10-1copies/μL,而cdPCR 为1.22×10-3copies/μL,说明cdPCR 比RT-qPCR 灵敏度高100 倍(表1)。
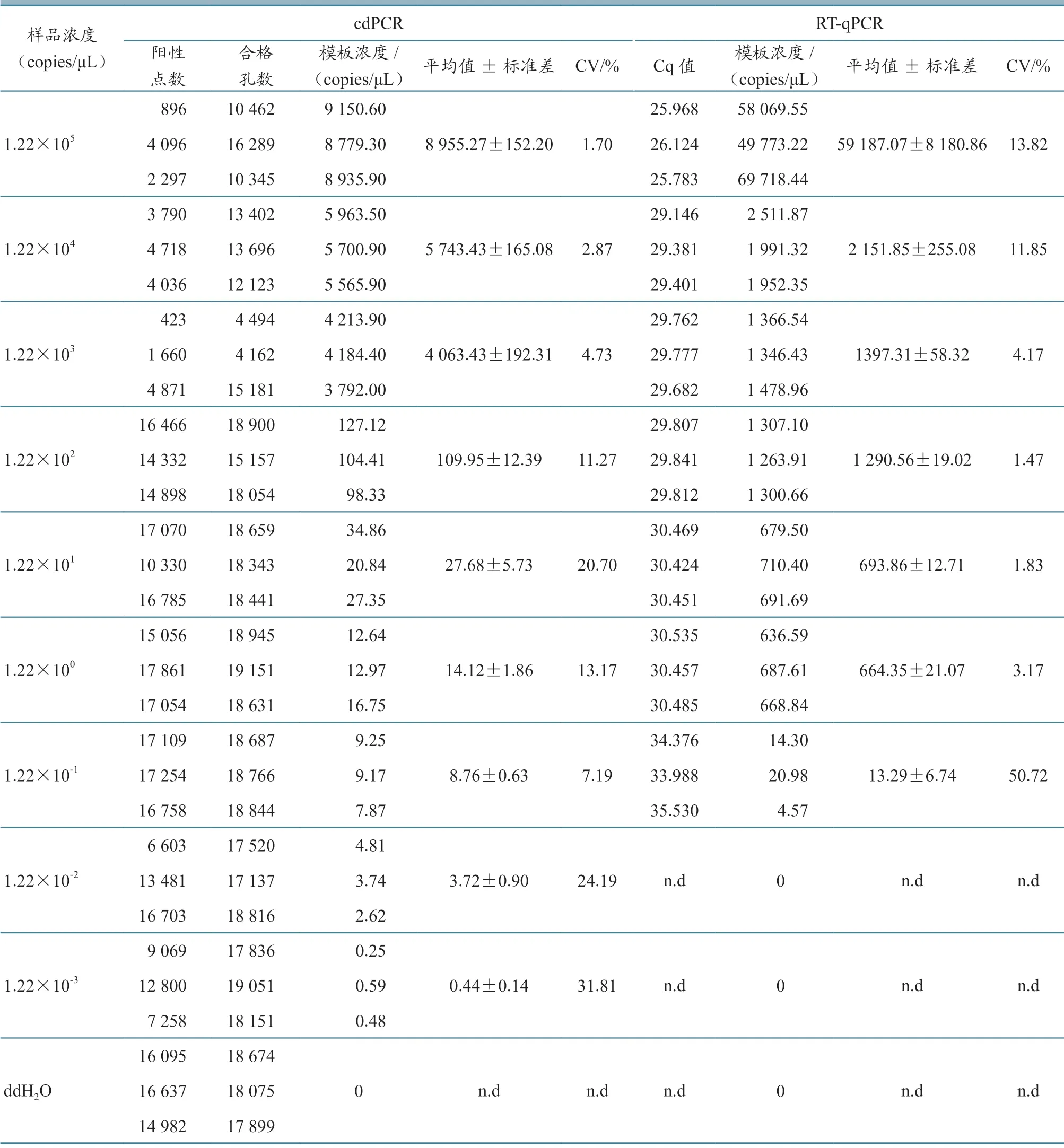
表1 cdPCR 和RT-qPCR 方法的敏感性试验比较结果
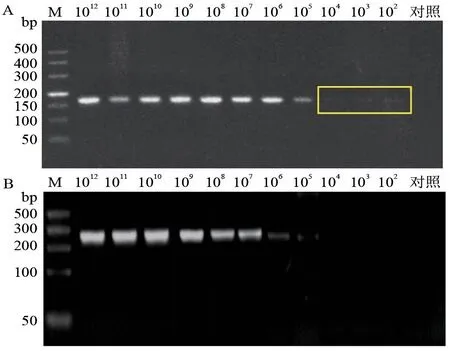
图4 不同引物的敏感性试验结果
2.5 特异性和重复性试验
特异性试验结果(表2、图5)显示,这两种方法均能特异性检测到PPRV,且与其他5 种病毒及阴性对照无交叉反应。对RT-qPCR 和cdPCR 检测的重复性进行评估,并结合定量结果变异系数(CV)来确定检测准确性。结果(表3)显示:cdPCR 的CV 范围为0.56%~10.73%,RT-qPCR 为3.11%~50.43%。这些结果表明,cdPCR 检测方法的重复性良好。

图5 RT-PCR 特异性试验结果
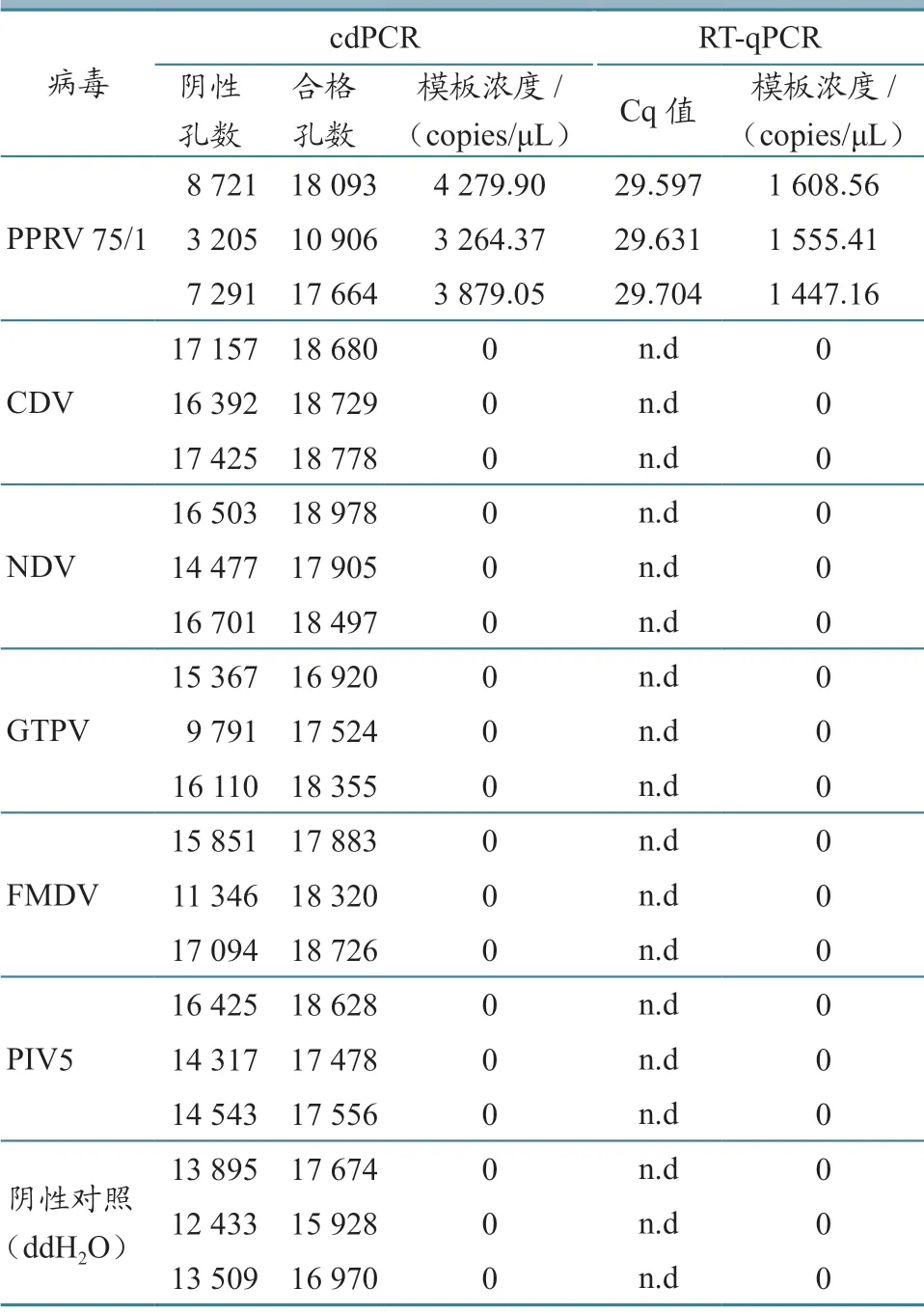
表2 cdPCR 和RT-qPCR 方法的特异性试验结果
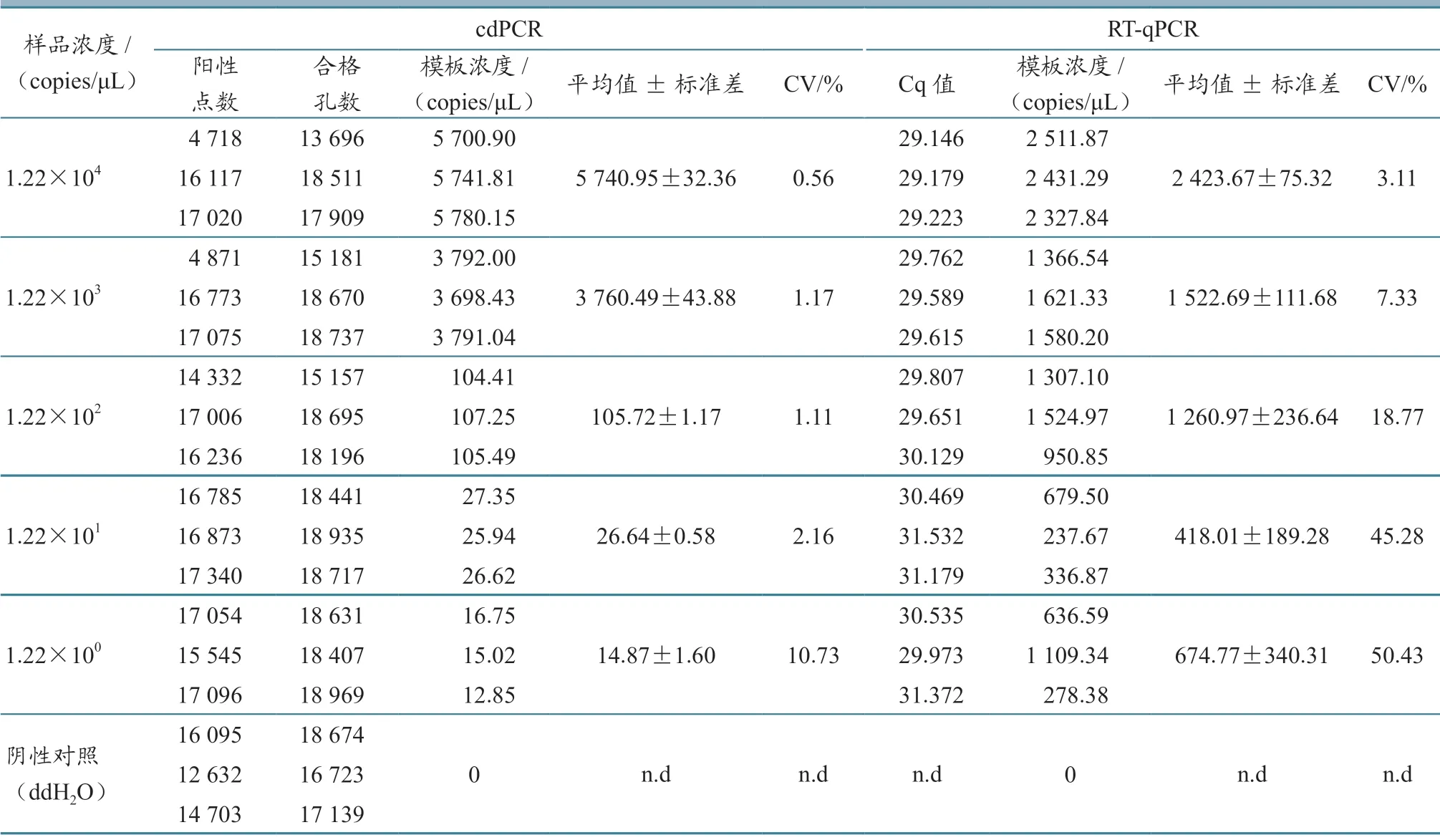
表3 cdPCR 和 RT-qPCR 的重复性试验结果
2.6 临床样品检测
采用RT-qPCR 和cdPCR 方法对47 份临床样品的检测结果(表4)显示,cdPCR 的检出率为25.5%,高于RT-qPCR 的17.0%。但两种方法的检测结果均显示,血液样品的检出率较低,肺、肠和淋巴结样品的检出率较高。
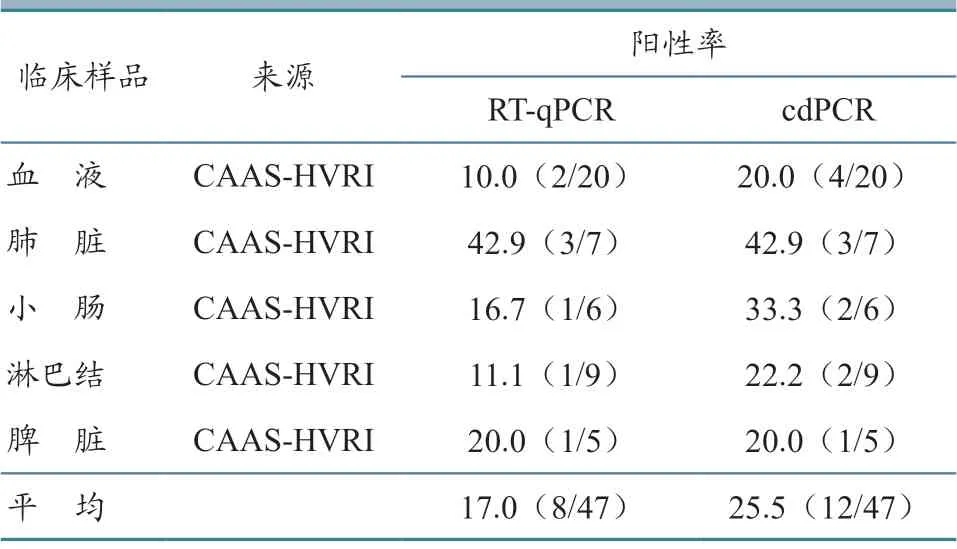
表4 临床样品的检测结果 %
3 讨论
PPRV 属于OIE 规定的须通报动物疫病,我国法定的一类动物疫病。2013—2016 年我国许多地区暴发了PPR,严重威胁我国动物疫病防疫安全[6]。我国对PPR 采取相应的控制和扑灭措施,使疫情得到有效控制,目前只有零星地区散发疫情。PPRV对热、干燥和紫外线比较敏感,因此在自然环境中的存活时间较短,但是在雨季和冬季会容易流行。PPRV 在我国传播的形式以输入性传播为主,集中在引种方面。引种时一旦没有做好及时的隔离和处置,就会有疫情地方性流行的风险。显然,早期快速和精准诊断在PPR 的防治方面将发挥着重要作用。
PCR 方法具有快速、敏感性高、特异性强等优点,但它需要后续电泳等处理,易出现假阳性和交叉污染[7]。相比传统PCR,cdPCR 具有更高的敏感性和特异性,同时缩短了检测时间,无须核酸电泳,结果直接读取,方便基层没有实验技术背景的工作人员操作。近年来 RT-qPCR 的应用已经改善了PPRV 核酸扩增方法[8]。该方法的灵敏度相对比常规RT-PCR 高10~100 倍,并且降低了污染风险。尽管RT-qPCR 核酸扩增方法在病原检测领域快速发展,但同时也伴随着一系列问题。例如:基于CT和ΔCT值的样品和/或标准品之间的相互依赖关系,在RT-qPCR 中,试验理想情况下需要在同一PCR 反应板上运行每个靶标的所有样品,最大限度地减少操作失误[9]。事实上,对于分布在多个位置之间的同一样品,要获得重复性好的试验数据一直是RT-qPCR 的一项重大挑战。尽管RT-qPCR 已广泛用于核酸定量,但它需要将未知物与标准品进行比较以获得定量信息。RT-qPCR基于荧光探针在PCR 的每个循环后监控扩增的模拟检测量,当样品的目标检测率低且差异基因表达水平低时,该技术检测能力受到限制。而相比RTqPCR,cdPCR 则不需建立标准曲线,直接给出靶序列的拷贝数,更加适合低拷贝核酸样品的检测。
本研究成功建立了一种cdPCR 检测方法用于PPR 诊断,显示出比OIE 推荐引物具有更高的灵敏度。cdPCR 相比RT-qPCR 在PPRV 特异性检测方面无明显差别,而在灵敏性方面比RT-qPCR 提高100 倍,且无需构建标准品和标准曲线,在相对病原浓度较少的样品中可发挥更高的灵敏度优势,同时样品处理也减少了“动手”所需的时间,并且芯片的密封可最大程度地减少非特异扩增子和其他核酸的污染。综上所述,本研究建立的cdPCR 检测方法具有特异性强、灵敏度高、重复性好的优点,在实际生产中可为预防PPR 早期流行提供一种快速有效的诊断和定量方法。
猜你喜欢
中国慈善家(2022年3期)2022-06-14
现代苏州(2022年9期)2022-05-26
快乐语文(2021年34期)2022-01-18
昆钢科技(2021年2期)2021-07-22
江西农业学报(2021年4期)2021-04-20
中国(俄文)(2020年8期)2020-11-23
职工法律天地(2018年12期)2018-01-22
地球物理学报(2016年8期)2016-09-29
西南医科大学学报(2015年1期)2015-08-22
中国当代医药(2015年9期)2015-03-01
