
膜上G蛋白偶联受体在运动影响骨代谢中的作用*
2022-03-31 02:11陈祥和陆鹏程周香香曾炘瑜
生物化学与生物物理进展 2022年3期
陈祥和 刘 波 陆鹏程 仇 啸 周香香 曾炘瑜
(扬州大学体育学院,扬州225127)
G 蛋白偶联受体(G protein couple receptors,GPCRs)的7次跨膜特征和晶体结构由2012年诺贝尔化学奖得主Robert J.Lefkowitz 和Brian K.Kobilka 发现[1]。GPCRs 是含有7 个跨膜α 螺旋的膜上受体,其跨过胞膜磷脂双分子层并响应胞外信号刺激,进而以G蛋白解离α亚基作为传导物触发细胞内部反应,介导正常或紊乱的生理反应[2]。G蛋白是膜内胞浆面的外周蛋白,与鸟嘌呤核苷酸结合并具有GTP酶(GTPase)活性,可将GTP水解,生成与膜受体偶联的异源三聚体——GDP[3]。细胞膜上GPCRs 可特异地与G 蛋白GTPase 区域结合,并形成GPCRs、G 蛋白及下游级联信号途径(包括腺苷酸环化酶、磷脂酶C、Ca2+通道、K+通道等)构成的信号转导途径[4],进而参与调控骨代谢、视觉传输、心率失常、癌变等。
骨中成骨细胞(osteoblast,OB)和破骨细胞(osteoclast,OC)相互影响、调控,分别主导骨形成代谢(以下简称骨形成)和骨吸收代谢(以下简称骨吸收),而GPCRs 可通过影响调控OB 和/或OC的信号途径、细胞因子等进而影响骨代谢[3,5]。敲除膜上G 蛋白偶联受体48 (G protein couple receptor 48,GPR48)、GPR54 后,小鼠骨中环磷酸 腺 苷 (cyclic adenosine monophosphate,cAMP)/蛋 白 激 酶A (protein kinase A,PKA)/Atf4、JNK/AP-1 等信号通路被抑制,导致分化产生的OB 减少但OC 却增多[6];而敲除另一受体亚型GPR40 后,OB 分化及骨形成增强且OC 分化被抑制,小鼠骨密度(bone mineral density,BMD)增加[7]。上述研究结果提示,GPCRs 在调控骨形成和/或骨吸收上扮演重要角色。甲状旁腺激素(parathyroid hormone,PTH)调控骨形成的分子机制与膜上G 蛋白及GPCRs 激活密切相关[8]。G 蛋白转导PTH 刺激进而激活靶蛋白β-arrestin/下游多囊蛋白1(polycystin-1,PC-1)/PDZ结合基序转录共激活因子(transcriptional co-activator with PDZ binding motif,TAZ)途径,以及Wnt3和Wnt7b信号刺激等来促进骨形成并抑制骨吸收[9-10]。后续研究关注小G蛋白在骨代谢中的调控作用,但因有研究发现G蛋白介导骨代谢是通过其异源三聚体而非小G 蛋白,所以有关其骨代谢调控作用尚存争议[3]。这可能与对小G蛋白研究尚不深入且相关研究集中于OB 和OC 分化有关。受限于G 蛋白分子结构单一及亚型数量少,其调控骨代谢关注度不高,研究较少。
运动是影响骨代谢的重要手段,而GPCRs(如GPR48等)作为力学刺激敏感蛋白在此过程中具有关键调控作用。目前,生命医学领域内有关GPCRs 调控骨代谢的研究较多,但运动通过调控GPCRs 进而影响骨代谢的研究较少,其作用机制及调控网络尚待揭示。基于此,本文将对GPCRs调控骨代谢及运动通过影响膜上GPCRs 进而影响骨代谢的相关研究进行综述并予以展望,以期为骨质疏松症等骨代谢疾病诊疗提供参考,并为运动医学领域内骨代谢研究提供一定的理论基础和新研究靶点。
1 GPCRs在骨代谢中的调控作用
1.1 GPCRs在骨形成中的调控作用
GPCRs 可与G 蛋白结合并调节其活性,介导cAMP、磷脂酰肌醇等信号途径[11]。其以单体、异源二聚体或寡聚体形式存在,刺激转导配体单一激活受体、寡聚复合物中相邻的其他受体来介导下游级联反应[12-13],调控阿尔茨海默病、高血压、心血管疾病、帕金森病等,亦可调控骨代谢[14-15]。OB膜上GPCRs 大量表达,且其信号支架蛋白GPCRs酶相互作用蛋白2 (GPCRs enzyme interaction protein 2,GⅠT2)可将外部刺激传入胞内调控骨形成代谢[6,16]。GPCRs 种类多,以下将详细阐述其对骨形成的介导调控。
1.1.1 GPR40
GPR40 属于7 次跨膜α 螺旋结构G 蛋白偶联孤儿受体,亦称游离脂肪酸受体1(free fatty acid receptor 1,FFAR1)[17],由碳链长度大于6 的中长链饱和及多不饱和脂肪酸(polyunsaturated fatty acids,PUFA)活化[18]。GPR40 不仅分布于胰腺细胞和神经细胞,也在OB膜上大量表达[4]。敲除GPR40后,Wnt途径关键因子β连环素(β-catenin)磷酸化被抑制,进而下调Runt 相关转录因子2(Runt-related transcription factor 2,Runx2)表达并作用于Sp7的PST序列区域,调节Fgfr2和Fgfr3来抑制OB 分化及骨形成[8]。OB 和脂肪细胞均由骨髓间充质干细胞(bone marrow mesenchymal stem cells,BMSCs)分化产生,而关键因子过氧化物酶体增殖物激活受体(peroxisome proliferatorsactivated receptor γ,PPARγ)受GPR40 调控,当GPR40 被敲除后会激活PPARγ 表达进而促进BMSCs 向 脂 肪 细 胞 分 化 并 抑 制OB 分 化[19]。GPR40 不仅调控脂质代谢,亦介导机体糖代谢。AMP依赖的蛋白激酶(adenosine 5'-monophosphate(AMP)-activated protein kinase,AMPK),作为介导糖代谢的关键蛋白,当抑制GPR40 后其表达下调,AMP/ATP 比率下降导致其对细胞能量代谢脱敏,内皮型一氧化氮合酶-一氧化氮(endothelial nitric oxide synthesis-nitric oxide,eNOS-NO)途径被抑制后Runx2 和锌指结构转录因子(osterix,Osx)去磷酸化不能与骨钙蛋白启动子区的成骨细胞特异性顺式作用元件2(osteoblast-specific cisacting element 2,OSE2)结合,抑制骨钙蛋白和细胞周期蛋白依赖性激酶抑制蛋白(cyclindependent kinase inhibitor,CKⅠ) 调 控 的OB 分化[20]。并且,GPR40 敲除后胰岛素分泌下降,胰岛素样生长因子(insulin-like growth factor 1,ⅠGF-1)B功能区N端16个氨基酸区与膜上胰岛素样生长因子1 受 体(insulin-like growth factor 1 receptor,ⅠGF-1R)结合,通过ⅠGF结合蛋白酪氨酸发出信号激酶受体,抑制Norrin及其调节的骨祖细胞增殖和向OB分化[21]。在探究罗格列酮对颅骨OB增殖及分化的影响时,发现GPR40 活化后磷酸化酪氨酸蛋白磷酸酶(Src-homology domain-2 phosphatase1,SHP-1) 并 抑 制 糖 原 合 成 酶 激 酶3β (glycogen synthase kinase 3β,GSK-3β),磷酸化SHP-1 促进激活经典Wnt 途径靶基因β-catenin;GSK-3β 被抑制后激活β-catenin,提高其在核内水平,促进前成骨细胞向OB分化[22]。调控OB分化及骨形成的信号途径呈网状结构分布,而GPR40作为膜上受体,是众多信号途径的“发起点”,除可介导众多信号途径外,对很多关键因子如血清骨钙素N端中分子片段(N-terminal middle molecular fragment of osteocalcin,N-MⅠD)、Ⅰ型胶原羧基端肽β 特殊序列(β-C-terminal telopeptide of type Ⅰcollagen,β-CTx)、T-Ⅰ型胶原氨基端前肽(T-procollagen type ⅠN-terminal propeptide,T-PⅠNP)、Dickkopf相关蛋白1(Dickkopf-related protein-1,DKK-1)和硬化蛋白(selerostin,SOST)等也具有重要调节作用,鉴于目前相关研究较少,其详细调控机制尚待揭示(图1,表1)。
1.1.2 GPR48
GPR48(亦称LGR4)属于A 家族糖蛋白激素受体,亦称富含亮氨酸的GPCRs[23]。以往研究中,GPR48 是未发现其配体的孤儿受体,而近来发现Rspondins(Rspo)家族蛋白中的Rspo1 与GPR48可通过LGRs家族胞外N端作用并开启经典Wnt信号通路,调控OB分化及骨形成[24],但对cAMP及Ca2+途径不显著。提示,GPR48可能存在其他配体来激活G 蛋白途径。研究显示,RANKL-/-小鼠表型与GPR48-/-小鼠类似,那么GPR48与RANKL可能存在一定关系。后续研究证实,除核因子κB 受体活化因子(receptor activator for nuclear factorκB,RANK)外,GPR48(LGR4)是核因子κB受体活化因子配体(receptor activator for nuclear factor-κB ligand,RANKL)的另一膜上受体,能通过胞外域(ECD)与RANK竞争性同RANKL结合,活化GSK-3β 和Gαq 途径,抑制核因子激活T细胞1(nuclear factor activates T cells 1,NFATc1)表达和活性从而介导OC 分化及骨吸收;LGR4(LGR4 CKO)小鼠显示OC过度活化(包括OC数量、表面积和体积增大)和骨质破坏增加[25]。这表明,Rspo1 和RANKL 是GPR48 配体,但关于其在骨形成中作用仍需进一步研究。
随着基因敲除技术发展,GPR48 调控骨形成作用引起学者关注,利用CRⅠSPR-Cas9 将其在OB上特异性敲除后发现,GPR48-/-小鼠和转录缺失(保留5%转录活性)小鼠骨组织出现BMD和骨组织形态结构退化,同时骨生长发育被阻滞[26-27]。分析其分子机制,GPR48 与GαS 结合后激活cAMP,促进PKA磷酸化并激活cAMP反应单元结合蛋白(cAMP-response element binding protein,CREB),后者是重要核蛋白,其调节启动子中具有cAMP 反 应 单 元(cAMP-response element,CRE) 的基因转录, 影响磷酸二酯酶(phosphodiesterase 4,PDE4)和胞外信号调节激酶(extracellular signal-regulated kinase,ERK)表达促进OB 分化及骨形成。经典Wnt 途径是调控OB 分化的关键途径,GPR48 活化后促进其靶基因β-catenin 磷酸化,入核后与T 细胞因子(T cell factor, TCF)/淋 巴 增 强 结 合 因 子(lymphoid enhancer factor,Lef)形成转录复合体,调节细胞周期蛋白D1(cell cycle protein D1,CyclinD1)、轴抑制因子2(axis inhibitor 2,Axin2)等靶基因表达来调节骨形成[28]。并且,Rspondins 通过其Furin 结构域与GPR48 结合可调控下游Wnt/平面细胞极化(planar cell polarity,PCP)途径[29];而敲除GPR48会抑制Wnt/PCP途径[30]。Luo等[25]发现GPR48 缺失导致骨长度变小,这与胚胎期骨形成延迟有关,与软骨成熟和增殖无关;GPR48 通过调节OB 分化、增殖及骨形成能力来达成这一功能。综上发现,GPR48调控骨形成由介导的OB分化及骨形成能力增强实现,与软骨细胞无关。但是,后续在研究PTHrP、PTH 和PTH/PTHrP 受体调控软骨发育时,发现GPR48 与PTHrP 作用,将外部刺激信号传递到胞内引发级联信号反应,促进原代软骨细胞3H-TdR 进入S-G2/M 期,使得成熟软骨细胞增殖。并且,二者偶联后亦可激活Hedgehog途径,Smo和Ptch1作为该途径关键跨膜蛋白,通过雌激素受体(estrogen receptor,ER)/Golgi 分泌途径进行C 端和N 端修饰,形成脂质态的具有高信号活性的信号分子物质,与Nkx3.2 和Sox9 形成自调节环路,调控软骨分化与增殖。另有研究发现,Hedgehog 途径激活后抑制OC 分化、融核及骨吸收。这表明,GPR48 促进OB 分化同时,亦可通过促进软骨细胞分化、增殖来促进骨形成,同时也会抑制OC 分化、融核及骨吸收(图1,表1)。
1.1.3 GPR54
GPR54 是Kiss1 蛋白受体,属于G 蛋白偶联受体视紫红质家族成员[31]。其包含1 197 bp 的开放阅读框,编码398 个氨基酸残基蛋白[32]。Kiss1 基因表达蛋白产物Kisspeptin,其具有一C端区域,为一个Arg-Phe-NHs 模体,由52 个氨基酸构成,RF-酰胺序列变为Arg-Tyr-NH2[33]。GPR54 具有介导能量代谢、性腺功能、肿瘤转移等生物学功能,有研究证实,GPR54 在OB 膜上大量表达。Kisspeptin 通 过GPR54 诱 导 蛋 白 激 酶C (protein kinase C,PKC)活化和磷酸酶抑制剂MCⅠP-1 表达,激活PKC/Runx2 通路关键因子Runx2,促进BMSCs 向OB 分 化[34],而GPR54 表 达 失 活 后β-catenin 去磷酸化,致使OB 功能紊乱[35]。OB 由BMSCs 分化产生,敲除GPR54 后Runx2、Osx 和C/EBPβ 表达下调并激活PPARγ,抑制成骨细胞前体细胞向成熟OB分化,导致细胞外基质成熟和基质矿化紊乱[36]。下丘脑-垂体-性腺(hypothalamuspituitary-gonad,HPG)轴不仅调控生殖系统发育和功能发挥,并且也可通过“脑-骨”串联影响骨代谢。HPG 轴调控睾酮分泌,下丘脑弓状核(arcuate nucleus,ARC)和前腹侧脑室周围核团(anterior ventral periventricular nucleus,AVPV)中的Kisspeptin/GPR54信号通路,可以调控下丘脑中促性腺激素释放激素(gonadotropin-releasing hormone,GnRH)的分泌、释放,影响性腺激素的分泌, GPR54 表达上调促进骨钙素(osteocalcin,OCN)分泌[37]。而OCN的活化形式羧化不全骨钙素(uncarboxylated osteocalcin,ucOCN) 激 活 磷 脂 酰 肌 醇 3-激 酶(phosphatidylinositide 3-kinases,PⅠ3K)后,上调靶基因蛋白激酶B(protein kinase B,Akt)表达并促进OB对Ⅰ型胶原蛋白、骨桥蛋白等有机质分泌,促进骨形成。并且,PⅠ3K激活后Akt磷酸化水平升高, OC 中Bax、 Bcl-2、 Caspase-3、 Cleavedcaspase-3 表达上调,促进OC 凋亡,降低OC 对骨组织的侵蚀[38]。上述研究显示,OB 和OC 均是GPR54 靶细胞,通过调控它们分化及功能发挥,可促进骨吸收(图1,表1)。
1.1.4 其他
研究发现,除上述阐述GPCRs 外,尚有其他GPCRs 可调控OB 分化及骨形成[39]。与ER 不同,GPR30 作为新发现的ER,其在骨中OB、OC 等细胞膜上大量表达[40]。当骨中GPR30 活化后,去卵巢大鼠骨转换率提升,BMD和生物力学性能升高,骨流失和骨组织形态微细结构退化改善[41]。在探究补骨脂素改善骨质疏松的研究中,小鼠骨中GPR30 被激活[42]后会活化ERK1/2 信号途径后并降低ANP 和β-MHC 表达,导致PSMD11 蛋白快速合成,进而介导雌激素调控MC3T3-E1分化产生的OB 线粒体自噬水平,保护OB 活性[43]。当利用GPR30 特异性激动剂G1 和特异性拮抗剂G15 来调控GPR30/GPER1 在全身表达时,发现介导骨形成关 键 途 径(cAMP/PKA/CREB、 MAPK (p38、JNK) 及PⅠ3K/Akt) 均 被 抑 制[44]。 这 表 明,GPR30 与以上途径存在密切级联反应,但有关GPR30介导以上途径进而调控OB分化或骨形成的相关研究尚待补充。GPR40和GPR120均是长链不饱和脂肪酸受体,将其敲除或转录缺失后,出现脂质代谢紊乱、胰岛素抵抗(insulin resistance,ⅠR)水平升高、大脑中海马区去甲肾上腺素水平升高等。而雌激素是治疗骨质疏松(ssteoporosis,OP)的最直接手段,BMSCs 膜上GPR40 受雌激素影响并调控OB分化,去卵巢大鼠骨中GPR40表达被抑制;将其敲除后,雌激素改善去卵巢GPR40-/-小鼠骨质疏松的作用缺失。这与GPR40 功能缺失后,Wnt/β-catenin 途径的关键因子β-catenin 去磷酸化,进而抑制OB分化及骨形成有关。另外,GPR40是通过Wnt/β-catenin 途径激活雌激素诱导OB 分化的内源性促进剂,该研究首次为GPR40 是通过Wnt/β-catenin 途径来正向调节骨形成提供了直接证据[45]。当用罗格列酮治疗2 型糖尿病(type 2 diabetic mellitus,T2DM)时,发现罗格列酮抑制OB增殖作用仅通过GPR40来实现,一旦GPR40功能缺失,其下游TLR4/JNK/NF-κB 途径将被抑制,那么GPR40 生物学作用将缺失[46]。长链多不饱和脂肪酸受体GPR120作为近来研究热点,在探究亚油酸抑制MC3T3-E1向OB分化及其骨形成能力时,发现亚油酸抑制MC3T3-E1 膜上GPR120 表达,下调β-arrestin2表达导致调控OB分化靶基因TAK1去磷酸化[47](图1,表1)。随着骨形成研究逐渐深入,相信更多GPCRs会被研究发现。
1.2 GPCRs在骨吸收中的调控作用
OC主导骨吸收,而OC的a3(116 ku)跨膜亚基电质子泵突变导致OC分化、融核紊乱和骨吸收异常升高[31]。膜上受体GPCRs 介导的信号途径或关键因子调控OC分化、融核及骨吸收能力。体外研究中,GPR1 表达上调后促进环氧化酶2(cyclooxygenase-2,COX-2)与RANKL 胞外结构域的TAAT 启动子原件作用,激活RANK 及下游NFATc1 等靶基因,促进OC 分化、融核[48],上调造血干细胞(hematopoietic stem cell,HSC)膜上GPR1受体表达后会抑制chemerin-CMKLR1-PPARγ途径,进而上调其靶基因adiponectin 并促进其向OC分化[49]。GPR30作为新型膜雌激素受体,其调控OC 分化与骨吸收,敲除GPR30 后通过上调ⅠGF-1表达促进雌性小鼠骨组织生长发育被显著抑制[50]。林紫微[51]研究发现,芍药苷的抗骨质疏松作用机制与膜上GPR30 表达上调后抑制CTSK、NFATc1、c-Fos、Dc-stamp 等调控的OC 分化及骨吸收能力密切相关。而作为脂肪酸受体的GPR40,当利用CRⅠSPR-Cas9技术将其在小鼠OC上特异性敲除后,NF-κB信号通路中的κB酶抑制剂(ⅠKKα/β)被抑制且下调OC 核内NFATc1 表达从而抑制OC 分化及其骨吸收能力[52]。其分子机制与GPR40-/-小鼠骨中NF-κB 途径及其靶基因NFATc1表达上调密切相关,当NFATc1 表达上调后促进破骨细胞前体向OC 分化[53]。并且,OC 与OB 之间并非独立存在,两者之间借Runx2来相互影响、调控。将OB 膜上GPR40 特异性敲除后,OB 分化被抑制的同时Runx2 表达下调,进而激活PⅠ3K/Akt途径,上调AP-1、c-myc、c-Src等靶基因表达,促进OC 分化[45]。然而,作为GPCRs 另一关键亚型的GPR48,将其全身敲除后骨生长发育被抑制,后续发现RANKL是其配体,可激活RANK及其介导的Gαq-GSK-3β途径,促进OC 分化及融核[25](图2,表2)。

Fig.1 Schematic diagram of the molecular mechanisms of GPCRs regulating bone formation图1 GPCRs调控骨形成的分子机制总结示意图

Table 1 Summary of molecular mechanisms of GPCRs regulating bone formation表1 GPCRs调控骨形成的分子机制汇总表
GPR54作为GPCRs的关键亚型,其缺失后OC封闭圈染色更亮,肌动蛋白环结构变大[36]。并且,GPR54 介导胞内信号途径(如:磷脂酶C(phospholipase C,PLC) β 异 构 体 的 活化 和磷脂酰肌醇-4,5-双磷酸(phosphatidylinositol-4,5-diphosphate,PⅠP2))的水解。而PⅠP2被水解成ⅠP3和DAG 两个第二信使后,引起下游Ca2+动员并作用于花生四烯酸释放,磷酸化ERK1/2 和p38 MAPK[32],下调NFATc1 表达[54];而p38MAPK 的活化会激活c-Fos途径及关键因子TRAF6表达,促进OC分化[55]。并且,ERK1/2与p38MAPK之间亦相互作用,p38MAPK抑制剂磷酸化ERK1/2,进而抑制OC 分化、融核[56]。而Kiss1 基因编码的肽类激素Kisspeptin-10(Kp-10)可刺激过表达GPR54 CHO细胞张力丝的形成,而此效应被外酶C3负向调控,此刺激效应通过激活小G蛋白Rho亚家族来进行调控。其C端结构域与c-Src SH3 结构域相结合后,抑制RANKL 蛋白表达和激活Kp-10 诱导的c-Src 磷酸化,形成RANK/avp3 复合物基团来激活Syk-Vav3-Rac1 信号通路,调控OC 分化和骨吸收[57](图2,表2)。受限于GPCRs 调控骨吸收代谢的相关研究较少,导致其相关分析有限且不深入,但确信GPCRs 将是未来生命医学领域的研究靶点和热点。

Fig.2 Schematic diagram of the mechanism of GPCR regulating bone resorption图2 GPCR调控骨吸收的分子机制总结示意图
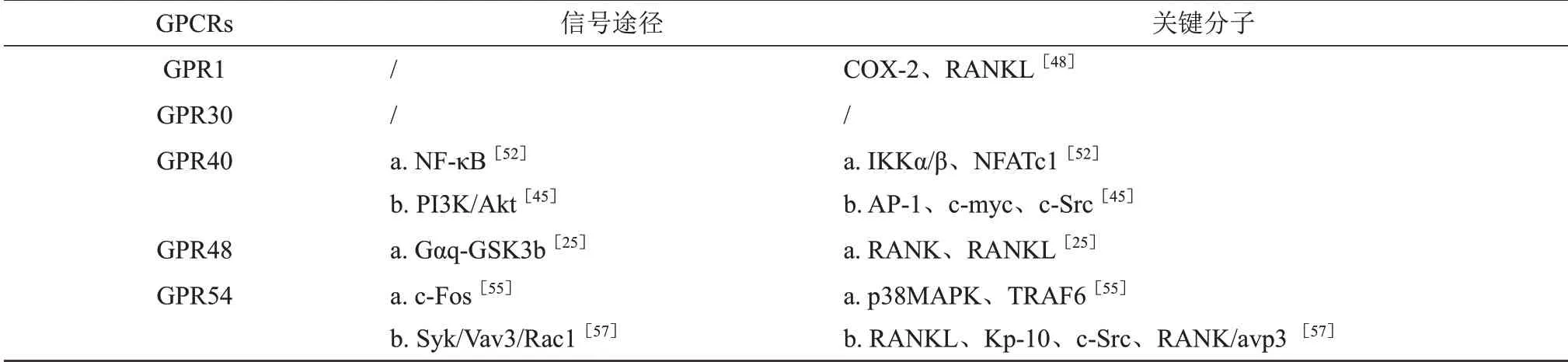
Table 2 Summary of molecular mechanisms of GPCRs regulating bone resorption表2 GPCRs调控骨吸收分子机制汇总表
2 GPCRs在运动影响骨代谢中的作用机制
骨以每年约占总骨量10%的速度进行更新,这种以“吸收-构建”为主要特征的骨动态平衡称为骨重建[58]。OB 和OC 的偶联平衡介导骨内环境稳态,研究证实,运动训练是改善骨内环境失衡导致代谢紊乱的关键因素。8周跑台训练使锰超氧化物歧化酶缺乏杂合小鼠骨形成加快,BMD 增加[59]。对比跑台训练,震动训练可显著上调大鼠股骨中OPG表达从而预防骨质疏松发生[60]。人体研究亦证实,16 周瑞士球稳定运动使得慢性腰痛患者BMD 增加[61]。综上,适度运动促进骨形成,改善骨代谢。
OB 和OC 膜上GPCRs 对力学刺激敏感,可将运动训练对骨产生的力学刺激传入膜内[62]。有关GPCRs 调控运动影响骨代谢的相关研究较少,其中以GPR48 研究为主。敲除GPR48 后,一方面cAMP/PKA/Atf4 信号通路活性被抑制,其靶基因Atf4表达下调,导致OCN、骨涎蛋白和Ⅰ型胶原蛋白等表达下调,降低骨形成[63],另一方面作为RANKL 受体,GPR48 能通过胞外域(ECD)与RANK 竞 争 性 同RANKL 结 合,活 化GSK-3β 和Gαq 途径进而抑制OC 分化[64]。在探究运动对T2DM OC 分化的影响时,发现下坡跑对T2DM 小鼠骨产生的直接作用力可激活骨中GPR48/RANKL通路,抑制RANK及其下游靶基因胞质活化T细胞核因 子c2 (nuclear factor of activated T cells c2,NFATc2)和组织蛋白酶K(cathepsin K,CTSK)表达, 入核后鞘氨醇磷酸酯(sphingosine 1-phosphate,S1P)失活,抑制破骨细胞前体细胞(osteoclast precursor cells,OCPs)向OC 分化及融核,且其作用效果优于游泳产生的间接作用力[65]。破骨细胞抑制因子(osteoclastogenesis inhibitory factor,OPG)/RANKL/RANK 分子轴可借助中枢因子肿瘤坏死因子受体相关因子6(TNF receptor associated factor 6,TRAF6)调节下游CN/NFAT和NF-κB途径,而运动借助OPG/RANKL/RANK分子轴和/或其下游CN/NFAT 和NF-κB 途径可抑制OC分化及骨吸收[66]。说明,GPR48 介导下游CN/NFAT 和NF-κB 途径是运动抑制OC 分化及骨吸收的分子机制。并且,该研究还对其分子机制网络进行了初探,认为TRAF6 介导的PⅠ3K/Akt、JNK/AP-1、p38MAPK、ERK1/2、LPS、CD40 等是该偶联网络的潜在机制[67]。GPR48 不仅调控OC 分化亦可介导OB 分化,并且其作为7 次跨膜受体可调控cAMP/PKA、Pitx2、Wnt/β-catenin 和BMPs 等信号途径并与其形成分子调控网络,从而影响OB分化及骨形成[68]。已有研究证实,运动训练可通过cAMP/PKA、Wnt/β-catenin 和BMPs 等信号途径调控OB分化。那么我们不禁思考,运动对细胞产生的力学刺激可能就是以膜上GPR48 为媒介,传入膜内来调控以上信号途径及成核结合因子(core-binding factor α1, Cbfα1) 、 PPARγ、
RANKL、OPG 等关键因子表达,实现运动促进OB分化。另一研究发现,在运动促进骨形成过程中,GPR48 可通过与TGF-β/Smads 途径调控OB 分化。并且,Norrin 作为GPR48 配体可上调其表达,磷酸化BMP-2 的Ser122 和Ser132 位点,使得Smad1/5/8 激活并与Smad4 形成磷酸化基团转移入核,上调靶基因表达来介导运动促进OB 分化[69](图3)。
GPR30 作为GPCRs 家族关键成员,其缺失后小鼠血液中骨吸收生化标志物CTX和股骨BMD下降[14]。而GPR30可调控骨形成关键蛋白BMP-6表达来发挥生物学作用。据此,当对骨施加运动力学刺激时,其可通过GPR30/骨形态发生蛋白(bone morphogenetic protein-6,BMP-6)途径来磷酸化Smad2、3、4并形成磷酸化复合基团,入核后调控BMSCs 向OB 的分化及骨形成(图3)。虽然膜上GPCRs 家族成员较多,但目前运动医学领域内有关运动通过膜上GPCRs 介导下游级联信号反应,进而调控OB和/或OC分化及骨代谢的相关研究尚待探究,这也将会是运动医学领域内研究骨细胞内环境稳态或骨代谢平衡分子调控机制的新热点和新靶点。
3 问题与展望
随着有关GPCRs 生物研究的不断深入,其在调控OB和/或OC分化及功能发挥进而影响骨代谢中的关键作用已广为认可。虽已有报道GPCRs 调控运动影响骨代谢,但相关研究较少。本研究拓展了GPCRs 的一个新功能,即其在运动影响OB 和/或OC分化及功能中的作用机制。国内外体育科学或运动医学领域有关GPCRs 调控骨代谢的专题研究较少,仍有很多问题亟待澄清,例如:a.还有哪些GPCRs?除已证实的GPR48 外,GPCRs 数量众多,还有哪些GPCRs 在运动改善骨代谢中发挥作用,并且相应的分子调控机制网络仍待揭示。b.选择何种运动方式及强度?前人研究已证实运动方式不同对骨代谢产生的效果迥异,GPCRs 在此过程中扮演何角色,且同一运动的强度不同对骨细胞产生的力学刺激差异较大,所以多大强度可激活GPCRs?c.是否调控OB 与OC 之间“crosstalk”?OB 与OC 之间可借Runx2 等关键因子相互交联,那么GPCRs 是否调控此过程且在运动改善骨代谢中是否也具有调控作用呢?明确以上问题,将有助于更加充分的熟知运动状态下GPCRs 调控骨代谢的分子机制网络,并为以后体育运动促进骨生长发育以及防治骨质疏松提供理论依据和研究靶点。

Fig.3 Schematic diagram of the mechanism of GPCRs in the effect of exercise on bone metabolism图3 GPCRs在运动影响骨代谢中的作用机制示意图
猜你喜欢
清华金融评论(2022年4期)2022-04-13
中国医药导报(2019年7期)2019-05-13
大科技·百科新说(2019年3期)2019-03-28
科教导刊·电子版(2018年9期)2018-06-07
分析化学(2018年2期)2018-03-02
分析化学(2017年12期)2017-12-25
古代文明(2014年1期)2014-02-23
文学教育·中旬版(2012年4期)2013-02-01
职业·下旬(2009年8期)2009-10-12
浙江中医杂志(2004年3期)2004-11-20
