
基于点云卷积神经网络的蛋白质柔性预测
2022-03-31 02:11张晓慧谷昊晟王知人
生物化学与生物物理进展 2022年3期
张晓慧 谷昊晟 王知人
(燕山大学理学院,秦皇岛066004)
随着后基因组时代的到来以及解析获得的蛋白质三维结构数据越来越多,蛋白质结构-功能关系研究成为结构生物学研究领域的重要科学问题,受到生命科学研究者的普遍关注。蛋白质是生物功能的主要实现者,生物体的一切生命活动几乎都离不开蛋白质的参与,蛋白质在基因表达调控、生物化学反应的催化、物质运输、信号传导、免疫防御、新陈代谢等生命过程中都发挥着关键性的作用。而蛋白质的各种生物学功能取决于其不同的空间结构以及特定的构象运动,蛋白质在不同的生理环境和功能状态下会呈现不同的空间构象,蛋白质生物学功能的实现有赖于其特定空间构象的转变[1],因此,蛋白质结构的动态性和柔性是蛋白质行使其生物功能的关键因素,基于蛋白质的三维结构预测其柔性运动特征,开发有效预测蛋白质柔性的数学模型和工具,有助于蛋白质生物学功能分子机制的研究,可以为药物设计和纳米分子器型的设计提供重要信息,具有重要的理论意义和应用价值。
关于分析和预测蛋白质柔性的问题很早就受到分子生物学家、计算生物学家等的高度关注。近几年,基于机器学习预测蛋白质柔性分析方法取得了良好的效果。2011 年Hwang 等[2]开发了一种基于支持向量机的机器学习策略预测了蛋白质表面loop区的柔性,使用蛋白质单体结构中loop 区残基的Ramachandran 角、晶体学B 因子和相对溶剂可及表面积三个特征,来区分蛋白质表面的柔性loop区和刚性loop 区,由表面loop 区的运动情况解释蛋白质-蛋白质结合时表面的构象变化。2017 年李海鸥[3]在蛋白质-配体对接骨架柔性以及全原子柔性的优化方面进行了研究,构建了一个基于多种深度学习模型的蛋白质二面角预测框架,精确地预测了蛋白质二面角,为蛋白质对接中骨架柔性的处理提供了一种非常有效的解决思路。同时,也提出了一种蛋白质整体结构重构的方法,使用了深度学习模型中的多层堆叠自动编码器模型,有效地避免了基于模板的传统蛋白质结构预测方法中的采样算法复杂、打分函数不准确的问题。2018 年Bramer等[4]结合多种机器学习模型和多尺度加权彩色图(MWCGs)方法,对蛋白质的B 因子进行了预测。该方法利用MWCGs来提取蛋白质结构的局部柔性特征,进而与蛋白质结构分辨率、重原子个数、结构解析的R实验值等蛋白质全局特征相结合,通过决策树、随机森林和卷积神经网络对大、中、小三组蛋白质数据集进行了训练和测试;结果表明,用机器学习方法得到的B 因子预测结果比用GNM 等传统方法得到的最小二乘拟合结果更准确。2020年Bemister-Buffington 等[5]通过基于图理论的ProFlex 方法获得蛋白质柔性分析,结合有效的机器学习模型对18个非活性和9个活性状态的蛋白质偶联受体(GPCRs)的跨膜螺旋和loop区进行了分析,有效识别了由配体触发的GPCRs 中配体结合位点的柔性转变。大量研究表明,机器学习方法是蛋白质结构和柔性特征分析和预测的有效工具,在蛋白质结构-功能关系研究中得到了广泛应用。
除了传统的机器学习方法,卷积神经网络(convolutional neural network,CNN)等深度学习模型在蛋白质结构-功能关系中也逐步得到了应用。CNN 模型在二维和三维图像识别领域取得了巨大成功和广泛应用,图像数据具有规则的数据结构形式,比如规则的二维网格或三维体素。而蛋白质结构数据是一种非规则的数据结构形式,为了构建适用于蛋白质体系的CNN 模型,现有的处理方法主要有两类:一类是人为地划分空间网格,将蛋白质结构数据转换为三维体素或图片集合的形式,进而采用图像处理领域常用的CNN 模型进行蛋白质体系的研究。2017年Jiménez等[6]从计算机视觉的角度将蛋白质结构视为三维图像,将三维空间离散成1Å × 1Å × 1Å 大小的体素网格,根据蛋白质中原子的体积,将所有原子映射到相应的网络上,同时,考虑了原子的7种特质(疏水性、芳香性、氢键受体或供体、带正或负电以及是否金属离子),将这些特征作为格点不同的特征通道,利用三维卷积模型,来预测蛋白质的活性位点。2020 年Wang等[7]开发了一种名为DOVE 的基于三维卷积操作的深度神经网络方法,以蛋白质-蛋白质相互作用界面为中心,设置203 Å3或403 Å3大小的盒子,将盒子进行网格划分,将蛋白质中的原子映射到不同的网格内,并将原子相互作用类型及其能量贡献作为神经网络的输入特征,利用三维卷积运算来预测蛋白质-蛋白质对接模式。另一类被大家广泛采用的方法是将蛋白质抽象为由大量的点和边所构成的分子图,图中的节点表示体系中的原子,边表示原子之间的化学键,进而通过设计特定的图卷积运算操作对蛋白质体系特征进行提取分析。2017 年Fout 等[8]将蛋白质结构抽象为节点和边所构成的图,评估了多个图卷积算子,通过对节点局部性质的卷积运算,来有效识别蛋白质-蛋白质相互作用界面,结果表明,这种方法预测的精度优于基于支持向量机的方法。2020年Mahmoud等[9]将蛋白质和配体抽象为由点和边所构成的图,对于蛋白质的图模型,每一个节点代表一个残基的Cα原子,节点的特征用残基的类型来表示,节点之间边的特征用两个残基Cα原子之间的距离来表示,对于配体的图模型,节点和边分别表示配体的重原子以及它们之间的共价连接。进而,利用图神经网络来预测蛋白质的Cα原子与配体重原子之间的距离。研究表明,相对于传统的分子对接方法,该模型对于蛋白质-配体结合模式预测的效率和准确性方面都有显著提高。然而,上述两类方法均存在一定的局限性和不足。在第一类方法中,需要将蛋白质体系内的所有原子映射到各自最近的网络上,这种映射过程会人为导致结构上的误差。同时,由于不同的蛋白质大小不一,对于体积比较小的蛋白质,存在大量的空网格,增加了很多不必要的计算。在第二类方法中,将蛋白质抽象为图的过程中,会人为丢失部分结构信息,比如原子之间的距离、相对位置等。
Qi等[10]提出了一种基于点云的卷积神经网络模型PointNet,在三维物体识别和分割研究中取得了非常好的效果,本文借鉴PointNet 模型的思想,将蛋白质体系视为大量原子所构成的点云,通过对点云的卷积运算来预测蛋白质结构的柔性。该方法直接将蛋白质体系中所有原子的三维坐标作为输入,不需要对蛋白质的空间结构进行人为预处理,保留了蛋白质结构中所有原子的位置信息。在卷积运算中,分别使用对称池化操作和空间变换网络[11]来处理点云的排列不变性和空间旋转不变性。与Qi 等的模型不同的是,不同的蛋白质结构所含有的原子数量不均一、数据尺寸不规则,为了实现网络的小批量训练方法,提出了一种新的批量化策略,使用大小不等的蛋白质小批量输入对网络进行迭代训练,并采用Pearson 相关系数作为模型训练的评价指标。同时,在网络的池化层和空间变换网络部分,采用了最大池化、平均池化串联的方法,有效避免一些极端情况发生,提升了网络的稳定性[12]。相对于大的蛋白质体系,小蛋白质的柔性更大,预测也更为困难,为此,本文选用小于60 个氨基酸的小蛋白质体系作为研究对象,在蛋白质数据库(protein data bank,PDB)中收集了243个非冗余蛋白质结构作为数据集,训练并测试了所搭建网络对蛋白质温度因子(B因子)[13]的预测效果,并与蛋白质柔性分析中广泛采用的高斯网络模型[14](Gaussian network model,GNM)的预测效果进行了比较。
1 材料与方法
1.1 数据集来源
本文使用的蛋白质结构数据来自于PDB 数据库(http://www.rcsb.org/),利用网站所提供的高级检索工具,按照如下标准搜集蛋白质结构文件数据,建立非冗余的小蛋白结构数据集:
a.蛋白质的残基数目范围在0~60;
b.蛋白质的结构通过X-射线晶体结构解析方法获得,并且分辨率在0.0~1.5 Å之间;
c.蛋白质结构中仅包含一条链;
d.所有蛋白质的序列同源性小于30%。
从满足上述标准的蛋白质结构数据中排除温度因子值全部相同和温度因子为0的蛋白质,最终剩余243个PDB文件。在这243个蛋白质体系中任意选取210个作为训练集,其余33个作为测试集。训练集的210个蛋白质PDB代码列在了附件表S1中,测试集的33个蛋白质PDB代码见表3。
1.2 模型的架构
本文借鉴了PointNet对三维点云进行局部结构识别和分割的思想,搭建了蛋白质柔性预测的卷积神经网络模型,对蛋白质的B 因子进行回归预测,具体网络结构如图1所示。

Fig.1 Network structure
1.2.1 网络算法的具体步骤
a.提取蛋白质氨基酸Cα原子的三维坐标作为输入,矩阵大小为N× 3,N代表蛋白质中氨基酸的数目,利用改进的空间变换网络T-net,预测出3× 3 的转换矩阵,将输入数据与转换矩阵相乘,获得蛋白质整体旋转后的坐标。
b.根据疏水性[15]和带电荷[16]情况将氨基酸分为4类,分别是非极性、极性不带电、极性带正电以及极性带负电。与氨基酸的20个种类相串联,形成一个24 维的特征向量,对提取的特征使用one-hot 编码将其映射到欧式空间,使模型能够更好地识别特征。将第一步变换后的坐标数据与24维特征进行串联,得到大小为N× 27的数据。
c.对第2步得到的数据通过三层卷积操作使其映射到高维空间,图中卷积层用Conv 来表示。第一层卷积(Conv1)的卷积核大小为1× 27,其余卷积核大小为1× 1,步长均为1,三层卷积核数目分别为64、128、128。
d.对第3步得到的数据再次通过T-net网络进行旋转。
e.将第4步旋转后的数据通过两层卷积操作映射到更高维的空间,卷积核均为1× 1,步长为1,卷积核数目分别为256、512,最后得到512维的高维特征。
f.对第5 步得到的高维特征进行对称池化操作,将15个蛋白质分开进行最大池化得到512维的全局特征向量。
g.在网络中添加跳跃连接[17]来建立层与层之间的关系,收集不同层中的局部特征,将第6步得到的全局特征复制N次,得到一个N× 512 的矩阵,将其与第3步和第5步得到的局部特征相串联,最终得到一个N× 1600的特征矩阵。
h.利 用 一 个 多 层 感 知 机[18]mlp(256,256,128)进行特征降维,其中三层卷积的卷积核大小均为1× 1,步长均为1,并运用dropout(本文设为0.8)来防止网络过拟合,最终得到网络预测的B因子值。
1.2.2 批量化策略
深度学习网络的优化常常采用小批量化策略,需要小批量的大小batch_size 参数所控制,代表每一次迭代所需的样本数,合适的batch_size 大小能够提高网络计算效率,减少训练一次全样本集所需的迭代次数,并且训练速度也可大大提高,同时batch_size 可以确定梯度的下降方向,使模型的收敛更加稳定。
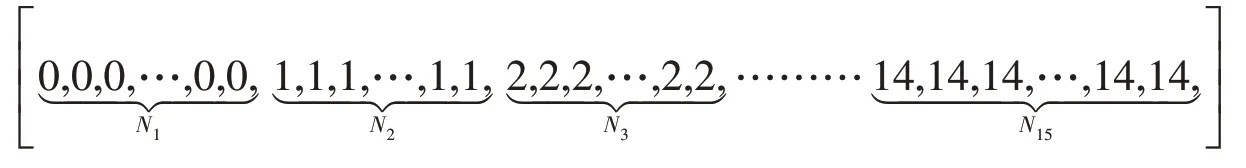
本网络使用的数据集是蛋白质结构,而蛋白质分子大小不一,会导致不同批次的小批量数据大小不规则,为了解决这一问题,本文提出了一种新的批量化策略,实现了输入蛋白质大小不等情形下的小批量迭代训练。本文设置的batch_size 大小为15,即同一批次放入15 个蛋白质数据。在网络的卷积运算过程中,当运行转换矩阵操作、使用对称池化操作来提取全局特征以及计算损失函数的时候是需要对同一批次的不同蛋白质体系进行区分。基于该网络结构,为了能够使15 个训练样本中的所有残基点一起进行训练,本研究将15 个样本进行串连,得到一个M× 3 的矩阵,M=N1+N2+N3+ …+N15,其中N1,N2…N15分别为15个样本中每个样本的残基个数,M为15 个样本残基个数的总和,将这个矩阵作为一次迭代的输入,用于网络第3 步与第5 步的卷积操作中,以提取每个残基的局部特征。
在应用变换矩阵、进行对称池化操作以及计算损失函数的时候,需将15 个蛋白质分开训练,因此本文使用tensorflow 框架下的数组拆分函数——tf.dynamic_partition 函数,将15 个样本进行分离。首先对M× 3 矩阵中的每一个残基都生成一个标签,这个标签代表这个残基所属哪一个蛋白质样本,最终得到一个M维的向量,形式如下:
接着使用数组拆分函数,操作效果如图2所示。
图中第一行表示每个元素的标签,第二行表示不同样本的点,第一行相同的数字表示它们属于同一个样本(图2),tf.dynamic_partition 函数可以通过标签将每一个样本都提取出来,即把15 个蛋白质分开,方便应用变换矩阵、进行对称池化操作以及计算损失函数,最终整体实现小批量化。

Fig.2 TF.Dynamic_Partition function renderings
1.2.3 模型的改进
为保持点云的置换不变形,在本文所建立的基于PointNet 的CNN 模型中,采用了最大池化操作来提取蛋白质体系的整体特征,最大池化操作与残基的排列次序无关,具有点云的置换不变性。除了最大池化外,平均池化也具有点云的置换不变性,为此,为了进一步提高网络的预测性能,在改进的模型中,同时考虑最大池化和平均池化,通过添加平均池化与最大池化串联的操作方法,将最大池化和平均池化提取的两种全局特征进行串联,得到包含更多蛋白质数据信息的全局特征向量,既保证了对蛋白质数据或特征排列的不变性,又增加了全局特征的信息量,使得模型更加稳定,具体结构如图3所示。

Fig.3 Improved symmetric pooling operation
1.2.4 评价指标
本文使用Pearson 相关系数作为评价指标,其计算表达式如下:
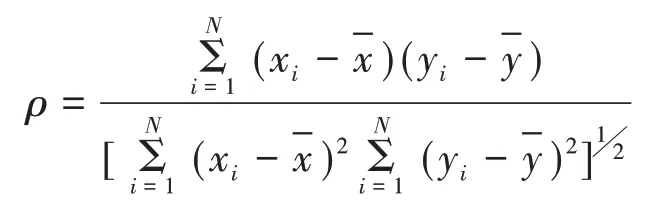
其中N代表蛋白质中氨基酸的个数,xi表示由本文网络预测出的B 因子值,i= 1,2,…。yi,i=1,2,…,i表示蛋白质PDB文件中的实验B 因子值,分别代表xi、yi的算术平均值。
1.2.5 损失函数
损失函数是深度学习中一个至关重要的结构,网络优化的过程,就是最小化损失函数的过程。Pearson相关系数的输出范围为[-1,+1],相关系数越大,则相关性越强,0 代表无相关性。因此,将1-ρ作为损失函数,ρ为Pearson 相关系数表达式,损失函数表达式为:

其中,N为向量中元素个数,xi与yi分别表示理论和实验B因子值,i= 1,2,…,分别代表xi、yi的算术平均值。
2 结果与讨论
2.1 模型参数的设置与优化
在深度学习中,调整超参数是优化网络的常用方法,通过观察本文网络的评价指标即Pearson 相关系数,可以判断当前网络处于什么样的状态,及时调整超参数可以科学有效的训练模型,节约大部分时间。针对蛋白质数据大小不等的特殊性,本文设计了新的小批量(mini-batch)优化策略,设置每一批的batch-size 为15,网络的优化过程采用动量(momentum)优化算法,训练轮数(epoch)为20,为防止过拟合,在最后利用多层感知机进行降维时设置丢弃率(dropout)为0.8。本文对学习率(learning rate)和动量两个超参数进行了调整优化,使得网络预测值与实验值的Pearson 相关系数达到最优,表1和表2显示了预测精度随超参数的变化情况。

Table 1 Optimization of the super-parameter learning rate
表1固定动量不变,调整学习率的大小,学习率是指在优化算法中更新网络权重的幅度大小,学习率过大可能会使模型不收敛,出现nan 的情况(nan 表示一些特殊数值,用于处理计算中出现的错误情况);学习率过小则导致模型收敛速度偏慢,训练时间较长。通常学习率有4个常用值,即0.000 1、0.001、0.01、0.1[19],本文在这4 个常用值附近进行调整优化获得最优的学习率参数值。由表1 可以看出当以0.000 1 作为初始值,优化到取0.000 3 时,Pearson 相关系数不再变化,则开始下一个取值点0.001 作为初始值进行优化。按照此规律调整至0.03 和0.1 时,相关系数出现了nan 的情况,这表明学习率太大导致模型不收敛。
表1 数据显示当学习率为0.002 时,相关系数达到最大,因此本文固定学习率为0.002 再进行动量的调整,结果如表2 所示。通常会尝试在0.9 到0.99 范围内设定动量值[19],并从中选择一个表现最佳值。由表2 可以看出当学习率取0.002、动量取0.97时,Pearson相关系数值达到最高。

Table 2 Optimization of the super-parameter momentum
2.2 与GNM方法的比较
GNM[20]是分析蛋白质结构固有柔性的有效方法,在蛋白质动力学性质分析以及蛋白质结构-功能关系研究中得到了广泛应用。在GNM 方法中,蛋白质的三维结构被简化为一个由大量节点和弹簧所构成的弹性网络,用蛋白质中氨基酸的Cα原子来代替每个氨基酸并以此为网络节点,当两个Cα原子之间距离小于截断半径时(本文取7.3 Å),节点之间用弹簧连接,模型中所有弹簧的弹性系数均相同。通过简正模式分析可以获得蛋白质体系的固有运动模式。大量的研究表明,目前GNM方法已经成为蛋白质B因子计算的主要理论方法之一,且预测结果较好,因此将本文方法的预测结果与GNM 计算方法的结果进行对比,验证该网络模型的有效性。在243 个数据集中随机选取210 个文件作为训练集,剩余33 个最为测试集。利用训练集对本文所搭建的网络模型以及改进的网络模型进行了训练,并利用训练好的模型对测试集中33 个蛋白质的B因子进行预测。大量的研究表明,蛋白质的柔性运动主要由其天然拓扑结构所决定[21]。同时,为了将本文模型的预测结果与GNM方法的计算结果进行比较,类似于GNM的做法,在本文模型中,对蛋白质体系进行了粗粒化处理,每一个氨基酸仅保留其Cα原子,其他原子均忽略。本文所构建的网络模型以及改进的网络模型的预测结果见表3和表4。作为对比,利用GNM方法对测试集中的33 个蛋白质体系的B 因子进行了计算。利用GNM方法进行计算时发现1ob4的第二个本征值为0,故排除。最终使用测试集中其余的32个蛋白质体系对三种模型的预测精度进行了对比(表3、4)。

Table 3 Pearson correlation coefficient of the B-factors for each protein in the test dataset predicted by our models compared with those predicted by GNM
表3 为32 个测试集中每个蛋白质B 因子的Pearson 相关系数,可以看出部分蛋白质基于PointNet模型和改进模型的预测结果要优于GNM,部分蛋白质比GNM差。经过对比测试集中各个蛋白质的结构发现,对于结构较为松散或N端和C端loop区较长的蛋白质来说,本文模型预测结果优于GNM,对于结构较为紧凑的部分蛋白质,本文模型略差于GNM。据统计,基于PointNet 模型和改进模型B因子预测结果高于GNM模型的蛋白质数量占比均为62.5%,改进模型预测结果高于基于PointNet 模型的蛋白质数量占比为53%。考虑到GNM 模型需要基于简正模分析理论,进行较复杂的物理计算,理论复杂,计算量大。而本文方法利用卷积神经网络直接提取蛋白质结构所固有的柔性特征,不需要复杂的理论分析,训练完成的网络模型可以方便的用于其他蛋白质体系的预测,计算简单快速,适用性好,并且预测效果略好于GNM方法。

Table 4 Mean Pearson correlation coefficient between the predicted and experimental B-factors for our proposed models compared with the results of GNM
表4 为32 个测试集中B 因子预测的平均Pearson相关系数,由表4可以看出相较于GNM网络,基于PointNet 模型的平均Pearson 相关系数提高了6.7%,改进后的模型平均Pearson相关系数提高了8.3%。由此可见,本文网络在预测蛋白质柔性方面效果较好。
2.3 大蜡螟丝蛋白酶抑制2 B因子的预测结果
为进一步说明本文网络的准确性,利用本文所提出的基于PointNet的卷积神经网络模型以及改进的模型对大蜡螟丝蛋白酶抑制2(PDB 代码4hgu)的B 因子进行预测,并与GNM 方法的计算结果进行比较。大蜡螟丝蛋白酶抑制2的三维结构见图4。该蛋白质整体是较为致密的球形结构,它由一个三股β片、一个α螺旋以及N端较长的loop结构所构成。本文所提出的基于PointNet的卷积神经网络模型以及改进的模型预测得到的B因子与实验B因子的Pearson 相关系数分别为0.80、0.85,而GNM 方法计算得到的B 因子与实验B 因子的Pearson 相关系数为0.69(图5)。

Fig.4 The tertiary structure of Galleria mellonella silk protease inhibitor 2

Fig.5 The B-factors of Galleria mellonella silk protease inhibitor 2 predicted by our PointNet-based model,the improved model and the GNM,respectively,compared with the experimental data
图5a为GNM模型预测得到的B因子与实验值的拟合曲线,可看出GNM模型对于大蜡螟丝蛋白酶抑制2 的N 端和C 端区域的柔性预测效果较差;图5b 为基于PointNet 的CNN 模型预测结果,图5c为改进模型的预测结果,可以看出基于PointNet的CNN 模型和改进模型的预测结果相差不大,二者略好于GNM网络模型。
2.4 无序蛋白质预测结果
GNM 模型对于结构紧密的蛋白质体系柔性预测效果较好,而对于结构松散的蛋白质体系,预测效果较差[22]。本文所构建的CNN模型对于结构紧密和松散的蛋白质体系均有较好的预测效果,尤其对于结构松散的蛋白质体系,本文模型的预测效果优于GNM模型。为了进一步验证这一点,本文利用所构建的模型对结构松散的天然无序蛋白质的B因子进行预测,并与GNM模型进行对比。从PDB数据库下载DisBind 网站[23]中无序蛋白质的PDB文件,去掉冗余蛋白以及在整个蛋白质中无序区域占比小于1%的蛋白质后,最终得到74个无序蛋白质,提取74 个蛋白质的无序区域作为数据集,按照本文方法随机选取60 个蛋白质作为训练集,14个作为测试集。训练集的60 个蛋白质列在了附件表S2 中,测试集的14 个蛋白质PDB 代码分别为
1jsu、1jwl、1l3l、1uad、2c1t、2f6a、3cxd、3hqr、3kz8、3m91、3pow、4jeh、4nm0、5hf7。利 用GNM模型对测试集的14个蛋白质进行B因子计算时,发现8 个蛋白质体系由于结构过于松散,GNM 计算得到的零本征值多于1 个,无法计算获得B因子结果。对于剩余的6个蛋白质体系,GNM模型和本文模型的预测结果列在了表5中。这里设置学习率为0.000 1、动量为0.97。
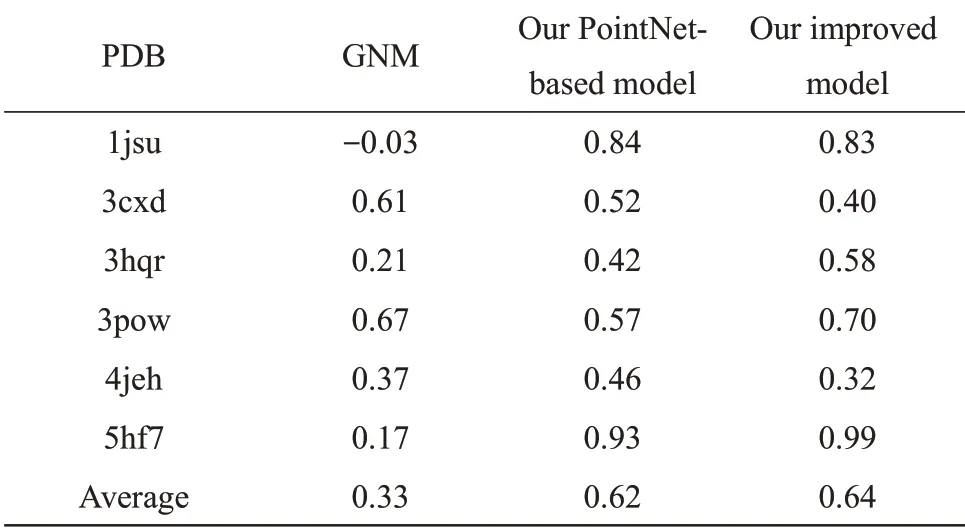
Table 5 Pearson correlation coefficient of the B-factors for each protein in the test dataset of disordered proteins predicted by our models compared with those predicted by GNM
表5为6个天然无序蛋白质B因子的Pearson相关系数以及平均Pearson 相关系数,可以看出相较于GNM模型,基于PointNet模型的平均Pearson相关系数提高了87.9%,改进后的模型平均Pearson相关系数提高了93.9%,由此可见,本文方法对结构较为松散的无序蛋白质预测效果明显优于GNM。
3 结 论
本文提出了一种基于PointNet网络的蛋白质柔性预测模型,此模型不需要对不规则的点云数据做任何处理,每个点仅由其三维坐标(x,y,z)表示,并作为输入数据直接传入网络,经过模型处理后即可输出蛋白质的B因子值,并计算出预测结果与实验结果的Pearson 相关系数。针对点云的排列不变性和空间旋转不变性,网络采用了对称池化操作和空间转换网络进行了有效处理,进而,改进的模型又在对称池化操作部分做了优化,有效控制了一些极端情况,使得预测结果更加准确。本文网络的基本架构非常简单,在保证一定准确率的基础上,减少了参数和计算量,有着较高的效率,提升了模型的稳定性。研究结果表明,在只考虑Cα原子的情况下,本文基于PointNet网络的模型和改进的模型得到的Pearson相关系数略好于广泛应用的GNM模型。尤其对于结构比较松散的天然无序蛋白质体系,本文方法预测结果明显优于GNM模型。
附件PⅠBB20200383_表S1-S2.pdf 见本文网络版(http://www.pibb.ac.cn或http://www.cnki.net)。
猜你喜欢
农业工程学报(2022年12期)2022-09-09
建材发展导向(2022年14期)2022-08-19
航天制造技术(2022年2期)2022-05-17
计算技术与自动化(2022年1期)2022-04-15
现代企业(2021年11期)2021-12-08
上海师范大学学报·自然科学版(2019年5期)2019-12-13
赢未来(2019年15期)2019-08-14
英美文学研究论丛(2018年2期)2018-08-27
数学学习与研究(2018年7期)2018-05-16
山东青年(2017年11期)2018-03-29
