
超高效液相色谱-串联质谱法同时检测毛发中112种合成大麻素类物质
2022-07-23 01:16张婷婷王优美2苏梦翔花镇东2
质谱学报 2022年4期
黄 钰,张婷婷,胡 爽,贾 薇,王优美2,,苏梦翔,花镇东2,
(1.中国药科大学药学院,南京 210009;2.国家禁毒委员会办公室中国药科大学禁毒关键技术联合实验室,北京 100193;3.公安部禁毒情报技术中心,毒品监测管控与禁毒关键技术公安部重点实验室,北京 100193;4. 北京警察学院,北京 102202)
合成大麻素(SCs)是新精神活性物质(NPS)的重要组成,该类物质能结合并激动生物体内的大麻素受体CB1和CB2,人体摄入后能产生类似吸食天然大麻的兴奋和致幻感。合成大麻素类物质的滥用于2004年在欧洲出现,其中以“Spice/K2”香料最为常见[1]。由于合成大麻素制造成本低、致幻效果强、新品种更新换代迅速,很快在世界范围内流行。《2021年世界毒品问题报告》指出[2]:合成大麻素仍然是目前世界上滥用最广泛的毒品,并且在过去十年里呈逐年递增的趋势。近年来,国内滥用合成大麻素制品的案例不断增多。为此,我国于2021年7月1日起,将合成大麻素类物质整类列入《非药用类麻醉药品和精神药品管制品种增补目录》,按照公告要求[3-4],符合图1所示7类化学结构通式的合成大麻素类物质均属于管制范围。为此,亟需建立相应的检测方法用于滥用人员的筛查识别。
注:R1代表取代或未取代的C3~C8烃基、取代或未取代的含有1~3个杂原子的杂环基、取代或未取代的含有1~3个杂原子的杂环基取代的甲基或乙基;R2代表氢或甲基或无任何原子;R3代表取代或未取代的C6~C10芳基、取代或未取代的C3~C10烃基、取代或未取代的含有1~3个杂原子的杂环基、取代或未取代的含有1~3个杂原子的杂环基取代的甲基或乙基;R4代表氢、取代或未取代的苯基、取代或未取代的苯甲基;R5代表取代或未取代的C3~C10烃基;X代表N或C;Y代表N或CH;Z代表O或NH或无任何原子图1 合成大麻素化学结构通式Fig.1 General skeleton structures of synthetic cannabinoids
目前,可用于毒品检测的生物样本包括尿液、血液、唾液和毛发。血液和尿液的检测窗口期较短,主要用于确定短期内的吸毒情况。然而,由于合成大麻素类物质极性较弱,血液和尿液中原体浓度极低,且代谢产物复杂,标准品不易获得,检测难度较大。相比之下,毛发样品具有易获取、易保存的优点,能够提供更长的检测窗口(长达数月),同时毛发中合成大麻素基本以原体形式存在,检测分析相对简单[5-6]。
目前,液相色谱-质谱联用技术已广泛应用于毒品的生物样本检测[7-9],其中关于合成大麻素定性、定量检测的研究逐年增加,但通常单个方法所涵盖的样品种类有限,尤其缺乏针对近期流行的吲唑酰胺和吲哚酰胺类合成大麻素的检测[10-12],而且进行高通量筛查时分析时间延长,检测效率降低[13-15]。
基于此,本研究拟利用超高效液相色谱-串联质谱(UPLC-MS/MS)法同时检测毛发中112种合成大麻素类物质,涵盖国家毒品实验室近年来监测发现存在非法制贩和滥用的所有合成大麻素品种,同时保证方法的高灵敏度,一次进样即可实现高通量的筛查及定量。
1 实验部分
1.1 仪器与装置
Triple QuadTM6500三重四极杆质谱仪:美国Sciex公司产品;ACQUITY UPLC I-Class超高效液相色谱仪:美国Waters公司产品;百万分之一电子天平:德国Sartorius公司产品;KQ-600DE型超声清洗器:昆山市超声仪器有限公司产品;冷冻研磨仪:北京万孚智能科技有限公司产品;Milli-Q advantage A10超纯水装置:德国Merck公司产品;具盖研磨管含氧化锆研磨球:北京柏丞科技有限公司产品。
1.2 材料与试剂
112种合成大麻素类物质标准品:均由公安部第三研究所提供,详细信息列于附表1(请登录《质谱学报》官网http:∥www.jcmss.com.cn下载);乙腈和甲醇:均为色谱纯,德国Merck公司产品;实验中所检测的毛发样品均为真实案件缴获。
1.3 样品制备
1.3.1标准溶液的制备 准确称取1 mg标准品,分别用甲醇溶解并定容至1 mL,得到1 g/L标准储备溶液,于-20 ℃密封避光储存。分别取适量的标准储备溶液,用甲醇稀释得到1 mg/L混合标准工作溶液,4 ℃下密封避光保存。系列混合标准工作溶液使用甲醇连续稀释得到,现用现配。
1.3.2样品提取 依次用适量的0.1%十二烷基硫酸钠(SDS)溶液、水、丙酮洗涤毛发样品,涡旋、振荡各5 min,置于通风橱中挥干。精确称取约20 mg毛发样品于具盖研磨管中,加入1 mL甲醇,于球磨机中研磨提取2 min,研磨提取液以14 000 r/min离心10 min,取适量上清液置于进样瓶中,供LC-MS检测。
1.4 实验条件
1.4.1色谱条件 条件1:色谱柱为ACQUITY UPLC BEH C18柱(2.1 mm×100 mm×1.7 μm);流动相为0.1%甲酸水溶液(A)和0.1%甲酸乙腈溶液(B);洗脱程序:0.0~9.0 min(5%~100%B),9.0~11.0 min(100%B),11.0~11.1 min(100%~5%B),11.1~13.0 min(5%B);流速0.4 mL/min;柱温40 ℃;进样量5 μL。
条件2:色谱柱为ACQUITY UPLC BEH C18柱(2.1 mm×100 mm×1.7 μm);流动相为0.1%甲酸水溶液(A)和0.1%甲酸乙腈溶液(B),采用50%B等度洗脱;流速0.4 mL/min;柱温40 ℃;进样量5 μL。
条件3:色谱柱为Kinetex F5 柱(3.0 mm×50 mm×2.6 μm);流动相为0.1%甲酸水溶液(A)和0.1%甲酸甲醇溶液(B),采用55%B等度洗脱;流速0.8 mL/min;柱温40 ℃;进样量5 μL。
1.4.2质谱条件 电喷雾离子源正离子模式(ESI+);多反应监测模式(Scheduled MRM);检测窗40 s;离子源温度500 ℃;电喷雾电压5 500 V;雾化气压强413.7 kPa;辅助加热气压强448.2 kPa;气帘气压强275.8 kPa;碰撞气为氮气;典型合成大麻素的信息列于表1。

表1 代表性合成大麻素的信息Table 1 Information of representative synthetic cannabinoids

续表1

续表1
2 结果与讨论
2.1 提取和进样溶剂的选择
目前,对毛发样品中毒品成分的提取方法主要有酸水解法、碱水解法和有机溶剂提取法[16-19]。酸水解法和碱水解法能将毛发完全消解,但由于合成大麻素类物质的水溶性不佳,消解后需要使用有机溶剂进行提取和浓缩[20-21],步骤较繁琐,不利于大批量样品的分析,且可能影响分子内含酯键合成大麻素的稳定性。甲醇是有机溶剂提取法常用的溶剂,对包括合成大麻素在内的各类毒品和新精神活性物质的提取效果良好[15,22],因此,本研究选择甲醇作为提取溶剂。
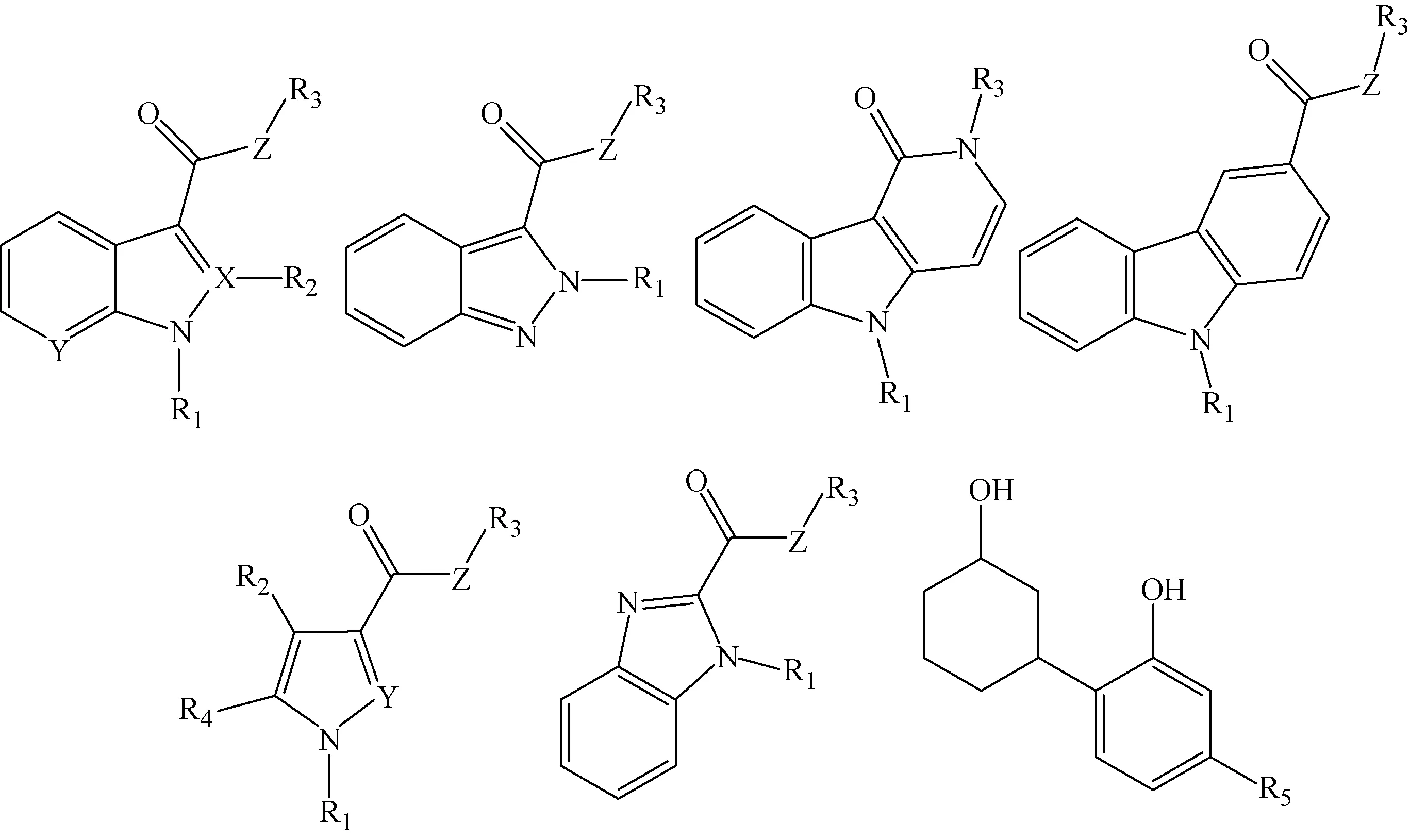
为避免色谱分离过程中的溶剂效应,在分析甲基苯丙胺等强极性毒品时,需要使用水对甲醇提取液进行稀释。本研究考察了甲醇-水溶液(1∶1,V/V)和纯甲醇溶液的进样情况,结果表明,所有物质在这2种条件下均具有良好的峰形,表明合成大麻素类物质极性较弱,受溶剂效应干扰较小。实验还发现,部分保留时间大于8 min的合成大麻素在甲醇溶剂进样时的响应更强,可能是由于这些物质在甲醇-水溶液中溶解度过低,有一部分被进样瓶吸附所致。以6种合成大麻素为例,对比图示于图2。为此,本实验最终选择甲醇提取液过滤后直接进样的方式。
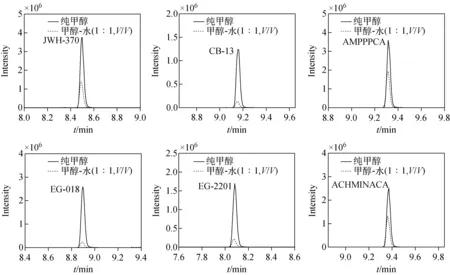
图2 不同溶剂体系中6种合成大麻素混合标准溶液(1 μg/L)的总离子流图Fig.2 Total ions chromatograms of standard solution for 6 synthetic cannabinoids (1 μg/L) in different solvent systems
2.2 色谱条件优化
由于不同骨架结构合成大麻素的极性存在较大差异,为保证所有目标物在合适的时间范围内出峰,优先采用梯度洗脱的方式进行色谱分离(1.4.1节中条件1),洗脱和平衡总时间为13 min,该条件下112种合成大麻素的出峰时间范围为3.58~9.32 min,色谱峰峰形良好,示于图3。由于分析目标物较多,不可避免地会出现多种物质洗脱时间接近的情况。即使在质谱分析过程中尽量选择特征性强的子离子,仍有一些物质的全部MRM通道存在交叉干扰,只能依靠洗脱时间的不同进行区分。如JWH-007和JWH-019互为同分异构体,二者在3个MRM通道的子离子质荷比完全相同,各通道峰面积的比例也高度相似,但二者的保留时间分别为7.98和8.19 min,差异大于2%。因此,可以通过色谱保留时间进行区分。
图3 112种合成大麻素的总离子流图(1 μg/L)Fig.3 Total ions chromatogram of 112 synthetic cannabinoids (1 μg/L)
此外,ADB-FUBINACA、APINACA-2H和5F-SDB-005这3种以吲唑为母核的物质,其所有MRM通道分别受相应的以吲哚为母核的ADB-FUBICA、APICA、5F-MN-18同位素峰的干扰,但可根据色谱保留时间的差异进行区分。
排除以上情况后,最终仅有FUB-PB-22和MDMB-FUBICA,AMB-FUBICA和MEP-FUBICA,以及5F-PB-22、5F-MDMB-PICA和5F-EMB-PICA这3组共7种物质的MRM通道完全相同。使用更平缓的洗脱梯度能改善分离效果,但分析时间将延长,不利于实际工作中大批量样品的筛查。为此,尝试建立独立的色谱洗脱程序用于以上物质的区分。当不改变色谱柱和流动相,采用1.4.1节的条件2时,分离情况示于图4a,FUB-PB-22和MDMB-FUBICA,以及5F-PB-22、5F-MDMB-PICA和5F-EMB-PICA可实现有效分离,但AMB-FUBICA和MEP-FUBICA仍被同时洗脱。进一步使用保留机理不同的五氟苯基固定相色谱柱,并使用含0.1%甲酸的甲醇作为有机相在55%比例下等度洗脱(1.4.1节条件3),AMB-FUBICA和MEP-FUBICA可实现有效分离,分离情况示于图4b。

注:a.1.4.1节条件2;b.1.4.1节条件3;1.MEP-FUBICA;2.AMB-FUBICA;3.5F-EMB-PICA;4.5F-MDMB-PICA;5.5F-PB-22;6.MDMB-FUBICA;7.FUB-PB-22图4 目标物的分离情况Fig.4 Separation of targets
根据以上实验结果,最终色谱洗脱条件确定为1个通用梯度洗脱方法和2个补充等度洗脱方法,当通用梯度洗脱方法筛查发现存在不能区分的物质时,可使用补充等度洗脱方法进行确证。鉴于这7种物质占目标物总数的比例仅为6.25%,可以预见,实际工作中需要使用补充方法的样品仅为少数,不会对筛查效率造成影响。
2.3 质谱条件优化
2.3.1离子对参数优化 首先,在流动注射模式下对112种合成大麻素的质谱参数进行优化。结果表明,绝大多数合成大麻素在ESI+模式下均可产生较高强度的[M+H]+准分子离子,在此基础上选择响应值高、特征性强的2~3个子离子建立MRM通道,并对去簇电压和碰撞能量进行优化。鉴于112种合成大麻素中存在大量分子质量接近、结构类似的物质,选择子离子时应尽量避开普遍存在的m/z144.0(吲哚甲酰离子)和m/z145.0(吲唑甲酰离子)等离子,以免产生不同物质之间的相互干扰。如ADB-BUTINACA和AB-PINACA的母离子均为m/z331.2,分别建立的3个MRM通道中有1个相同的子离子m/z286.2,其余2个互不相同,可有效区分。5F-AMB-PICA和4F-MDMB-BUTICA,ADB-BINACA和5Cl-AB-PINACA,5F-AMB和4F-MDMB-BUTINACA这3对物质具有类似情况。此外,优化过程中发现,NM-2201分子中的酯键稳定性较差,易发生源内裂解,导致碎片离子m/z232.1的强度远高于准分子离子m/z376.2,且m/z376.2碰撞诱导解离后得到的子离子也以m/z232.1为主。因此,除m/z376.2/232.1通道外,其余2个MRM通道的选择以m/z232.1为母离子,能有效提高检测灵敏度。最终优化得到的112种合成大麻素的离子对参数列于附表1。
2.3.2离子源参数优化 离子源参数的选择会对离子化效率及灵敏度产生影响。本实验以1 μg/L混合标准溶液为样品,对离子源温度、电喷雾电压、雾化气压强及辅助加热气压强等质谱参数进行优化,以化合物定量离子通道的最优灵敏度,确定最佳参数。实验结果表明,大部分目标物在离子源温度500 ℃和电喷雾电压5 500 V下具有较好的灵敏度,13种代表性物质的离子源参数优化结果示于图5,而雾化气和辅助加热气无显著影响。

注:a.不同离子源温度;b.不同电喷雾电压图5 13种代表性合成大麻素的离子源参数优化结果Fig.5 Optimization results of ion source parameters for 13 representative synthetic cannabinoids
2.4 方法学验证
2.4.1检出限、定量限和线性范围 取空白毛发按照1.3.2节方法处理,得到空白基质溶液,并以此配制成一系列空白添加溶液进行测试,分别以信噪比(S/N)>3和S/N>10确定检出限(LODs)和定量限(LOQs),线性相关系数r≥0.999 0作为线性范围。结果表明,112种合成大麻素的检出限范围为0.2~1 ng/g,定量限范围为0.5~5 ng/g;AB-FUBINACA、5F-ADBICA、ADB-PINACA、5F-ADB-PINACA、APINACA-2H、EMB-FUBINACA、5F-BTP7AIC、5F-MN-24及EDMB-PINACA在1~100 ng/g浓度范围内的线性关系良好;JWH-250、A-834,735、A-796,260、CUMYL-PEGACLONE及5F-CUMYL-P7AICA在2.5~250 ng/g浓度范围内的线性关系良好;5F-APINAC在5~500 ng/g浓度范围内的线性关系良好;其余97种合成大麻素在0.5~100 ng/g浓度范围内具有良好的线性关系,表明该方法具有较高灵敏度,可满足毛发中痕量目标物的检测要求。各目标物的检出限、线性范围及相关系数列于附表2。
2.4.2基质效应和提取回收率 向空白毛发中添加1、5、25、100 ng/g的112种合成大麻素类物质,并按照1.3.2节方法处理,测得目标物峰面积为A;用甲醇溶剂配制相应浓度的混合标准溶液,测得目标物峰面积为B;取空白毛发按照1.3.2节方法处理,得到空白基质溶液,配制相应浓度的空白添加溶液,测得峰面积为C。提取回收率=A/C×100%,基质效应(ME)=C/B×100%。根据实验结果,4个添加浓度水平下,112种合成大麻素的基质效应为81.0%~117.8%,无明显的基质抑制和基质增强作用,提取回收率为80.4%~119.7%,表明本方法的定量分析结果具有准确性和可靠性。各目标物的回收率和基质效应结果列于附表2。
2.4.3日间和日内精密度 向空白毛发中添加1、5、25、100 ng/g的112种合成大麻素类物质,按照1.3.2节方法处理并进样分析,每个加标水平在1天内平行测定6次,计算相对标准偏差(RSD),得到日内精密度(intra-RSD);连续重复6天,得到日间精密度(inter-RSD)。结果表明,112种合成大麻素的日内精密度为0.4%~7.7%,日间精密度为1.0%~10.6%,表明该方法的重复性良好,对毛发中多种合成大麻素的分析具有可靠性和实用性。各目标物的精密度结果列于附表2。

表2 实际样品检测结果Table 2 Test results of actual cases
2.4.4稳定性 向空白毛发中添加1、5、25、100 ng/g的112种合成大麻素类物质,按照1.3.2节方法处理,在室温下2 h内重复进样6次提取液,放置4 ℃冰箱冷藏6天后再次重复进样6次,比较前后2次测定响应值的平均值。结果表明,放置6天后,5F-BTP7AIC的响应值显著降低,4个加标水平为冷藏前的0%~27.7%,这可能是由于该物质中酯键与含有氮原子的三唑环相连易发生分子内催化酯水解;吲唑酰胺及吲哚酰胺类合成大麻素响应降至冷藏前的50%~70%,表明该类结构物质的稳定性欠佳;其余大部分合成大麻素响应降为冷藏前70%~95%。以上结果表明,毛发样品处理后应立即检测,避免长期放置,以保证实验结果的准确性和可靠性。
2.5 实际样品分析
利用本方法检测某地公安部门采集的15份疑似吸食合成大麻素人员的毛发样品,检出4CN-CUMYL-BUTINACA、MDMB-4en-PINACA、4F-MDMB-BUTICA、ADB-BUTINACA、5F-MDMB-PICA等5种合成大麻素类物质,含量为1.4~48.2 ng/g,详细结果列于表2。其中2号检材检出了所有5种物质,各物质的MRM通道均未出现干扰峰,结果示于图6,表明本方法所选离子对的特异性好、抗干扰能力强。合成大麻素类物质种类繁多,更新换代迅速,且常见多种混合使用的情况,以上5种物质均为近半年来国家毒品实验室在缴获样品中检出频率较高的品种。在实际样品检测中,筛查方法应纳入尽量多的目标物以避免漏检,本方法涵盖了国内已发现的所有合成大麻素类物质,同时可根据流行趋势监测结果增加新发现的物质,具有较高的实用性和扩展性。

图6 2号检材中检出的5种合成大麻素MRM色谱图Fig.6 Chromatograms of five synthetic cannabinoids detected in the sample 2
3 结论
本研究建立了UPLC-MS/MS法同时检测毛发中112种合成大麻素类物质,并对提取回收率、基质效应、日内和日间精密度、检出限、定量限、线性范围等参数进行考察,可成功用于实际毛发样品的检测。本研究实现了对毛发中目前国内已发现的所有合成大麻素类物质的同时定性和定量分析,能有效区分化学结构和性质接近的类似物,前处理方法简便、分析耗时短,可用于实际工作中大批量样品的检测。
猜你喜欢
食品安全导刊(2021年21期)2021-08-30
昆明医科大学学报(2021年2期)2021-03-29
法制博览(2020年27期)2020-11-30
中国生殖健康(2019年11期)2019-01-07
科技与创新(2018年21期)2018-11-30
青少年科技博览(中学版)(2017年5期)2018-02-28
儿童故事画报·发现号趣味百科(2017年1期)2017-06-01
华声(2016年20期)2016-11-19
大自然探索(2015年10期)2015-09-10
小火炬·阅读作文(2014年5期)2015-04-07
