
WGCNA共表达网络显示CDC45和MCM2与宫颈癌预后相关
2022-08-27 06:24李昊晨张子威李素婷
承德医学院学报 2022年4期
李昊晨,耿 静,张子威,李素婷
(1.承德医学院研究生院,河北承德 067000;2.承德医学院中药学系;3.承德医学院基础医学院)
宫颈癌是全球妇女排名第四的常患癌症,据估计2018年有57万个新发病例,全球31.1万名妇女死于宫颈癌,其中90%发生在低收入和中等收入国家[1]。在中国,宫颈癌是最常见的女性生殖系统癌症[2]。2013年,年龄标准化发病率为每100000人10.3例[3],5年相对存活率为45.4%[4]。近年来,全球女性宫颈癌的平均发病率有所下降,但是预计到2030年,每年被确诊的患者将超过70万例[5]。随着人均寿命的延长,诊断、治疗方法的发展,宫颈癌的5年生存率较高,预后研究却相对较少[6]。从分子层面找出与宫颈癌发生发展及转归相关的基因,对该疾病相关诊疗具有重大意义。加权基因共表达网络(weighted correlation network analysis,WGCNA)分析是通过构建无尺度基因共表达网络,从而分析各个基因模块以及临床症状与基因模块间的相关联系的方法[7]。该方法在胃癌[8,9]、口腔鳞状细胞癌[10]、肝癌[11]等疾病的研究中成功筛选出疾病相关基因。本研究采用WGCNA分析的方法对宫颈癌细胞进行分析并筛选关键基因。
1 材料和方法
1.1 临床数据和表达数据的获得及预处理
从GEO(https://www.ncbi.nlm.nih.gov/geo/)数据库获取登录号为GSE63514的宫颈癌基因表达数据。使用R对数据进行初步的整理后获得癌症基因表达矩阵。然后根据
[HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0
Array平台注释信息,将原始数据中的Ensembl ID转换成与其对应的基因名。
1.2 差异基因的筛选
使用R(https://www.r-project.org/)的“edgeR”包对数据进行处理,对有多个表达数据的基因求平均值。获得的表达矩阵用CPM矫正后再将所有样品里表达量都很低的基因从表达矩阵中剔除。然后用“limma”包对表达矩阵进行差异分析[12]。差异表达基因的筛选条件设定为logFC>0.05和adj.P.Val>0.05。用FDR法对差异分析的结果进行校正。矫正后最终得到的差异基因用于后续分析。
1.3 共表达网络的构建和模块保守性分析
在R中使用软件包“WGCNA”构建无尺度基因共表达网络[7,13]。检查确认表达矩阵数据中无缺失值后首先对样品进行聚类,将与其他样品差异过大的离群样品删除。选出合适的软阈值是构建出的网络更符合无尺度网络标准。计算各基因间的邻接值并构建出拓扑重叠矩阵。根据之前选定的软阈值,将所有基因划分为至少包含50个基因的模块,用动态树剪切法将相关性小于0.25的模块合并,将所有基因划分为数个模块,分别计算每个模块和正常组织与癌组织的相关性,挑选出相关性绝对值最大的模块并用分析其保守性[14]。经分析该模块的Preservationc Zsumary评分高于10分,则认为该模块基因保守性较高,具有研究意义,可以进行后续分析。取该模块基因和差异表达基因以及TCGA(https://portal.gdc.cancer.gov/)获取的CESC表达数据的WGCNA结果和差异分析结果的交集。则交集内的基因的表达量变化与组织的癌变相关性最大。
1.4 蛋白互作网络构建及核心基因筛选
使用string(https://www.string-db.org/)在线将之前获得的交集基因构建成PPI共表达网络[15],互作关系评分设定为0.9然后输出PPI网络。使用Cytoscape(https://cytoscape.org/)打开在string构建的蛋白互作网络,使用Cytoscape内的“CytoHubba”包从蛋白互作网络中筛选出模块中的前十个核心基因[16]。
1.5 GO富集分析和KEGG富集分析
使用R包“org.Hs.eg.db”将基因名转化为entrezlID。然后使用R包“clusterProfiler”,“org.Hs.eg.db”,“enrichplot”和“ggplot2”包进行基因本体论分析GO(Gene Ontology)[17]和京都基因和基因组百科全书富集分析KEGG富集分析(Kyoto Encyclopedia of Genes and Genomes)[18]。
1.6 转录因子调控网络构建
使用DAVID(https://david.ncifcrf.gov/)在线将模块基因富集到调控因子上。使用perl脚本整理转录因子调控网络并使用Cytoscape绘制可视化的共表达网络图。
1.7 生存分析和无病生存期分析
通过GEPIA2(http://gepia2.cancer-pku.cn/#index)在线进行生存分析[19]。从结果中筛选出P.Value<0.05的基因,这些差异表达的基因被认为具有统计学意义。然后再用Oncomine(https://www.oncomine.org/resource/login.html)数据库对差异表达具有显著统计学意义的基因进行验证。
1.8 临床相关性分析
在GSE63514(https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE63514)表达数据中有宫颈上皮内肿瘤的低度病变、中度病变和高度病变以及宫颈鳞状上皮癌几种状态。结合核心基因的表达数据判断核心基因与这几种性状间的相关性。
2 结果
2.1 差异表达基因筛选
下载登录号为GSE63514的宫颈癌基因表达数据中共128例样本,其中24例正常样本,104例肿瘤样本。肿瘤样本中包括40例高分化度样本,22例中分化度样本,14例中分化度样本。为了筛选出可能与癌症相关联的基因,我们对所有基因进行了差异分析,结果显示,GSE63514数据集21654个基因中,431个为上调基因,360个为下调基因,共计791个存在差异表达的基因。绘制差异基因的火山图(图1A),挑选差异最显著的50个基因和下调最显著的50个基因绘制热图(图1B)。
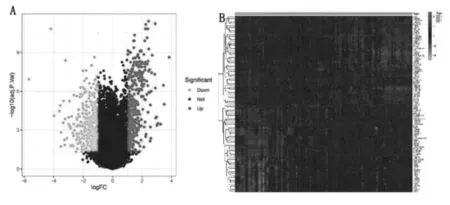
图1 基因表达差异分析结果
2.2 基因共表达网络构建及模块保守性分析
为了从791个表达存在差异的基因中筛选出和癌症相关性更强的基因,我们将矫正过后的基因表达数据用于WGCNA分析。先将基因进行聚类(图2A)。分析结果显示当选择邻接矩阵的软阈值为4时网络中的基因之间链接最符合无尺度网络分布(图2B)。所以设定power值为4进而构建出18个基因模块以及分层聚类树。然后以相似系数为0.25为标准对聚类树进行剪切。最终获得17个基因模块(图2C)。在这些模块中名为MEblack的黑色模块与宫颈癌的相关性最强(图2D)。说明MEblack模块中的基因和癌症相关性更强。对所有模块进行保守性分析(图2E),结果显示MEblack模块保守性大于10,说明该模块内基因在肿瘤细胞中表达状况稳定,因此该模块具有进一步分析的意义。为了验证WGCNA分析的结果,下载TCGA数据库中CESC的表达数据,其中包括3例正常样本和306例肿瘤样本。一同进行差异分析和WGCNA分析后取分析结果的交集。最后共同筛选出125个差异基因。这些基因经过数据集GSE63514和TCGA的CESC表达数据双重验证,可能为与宫颈癌高度相关的基因。
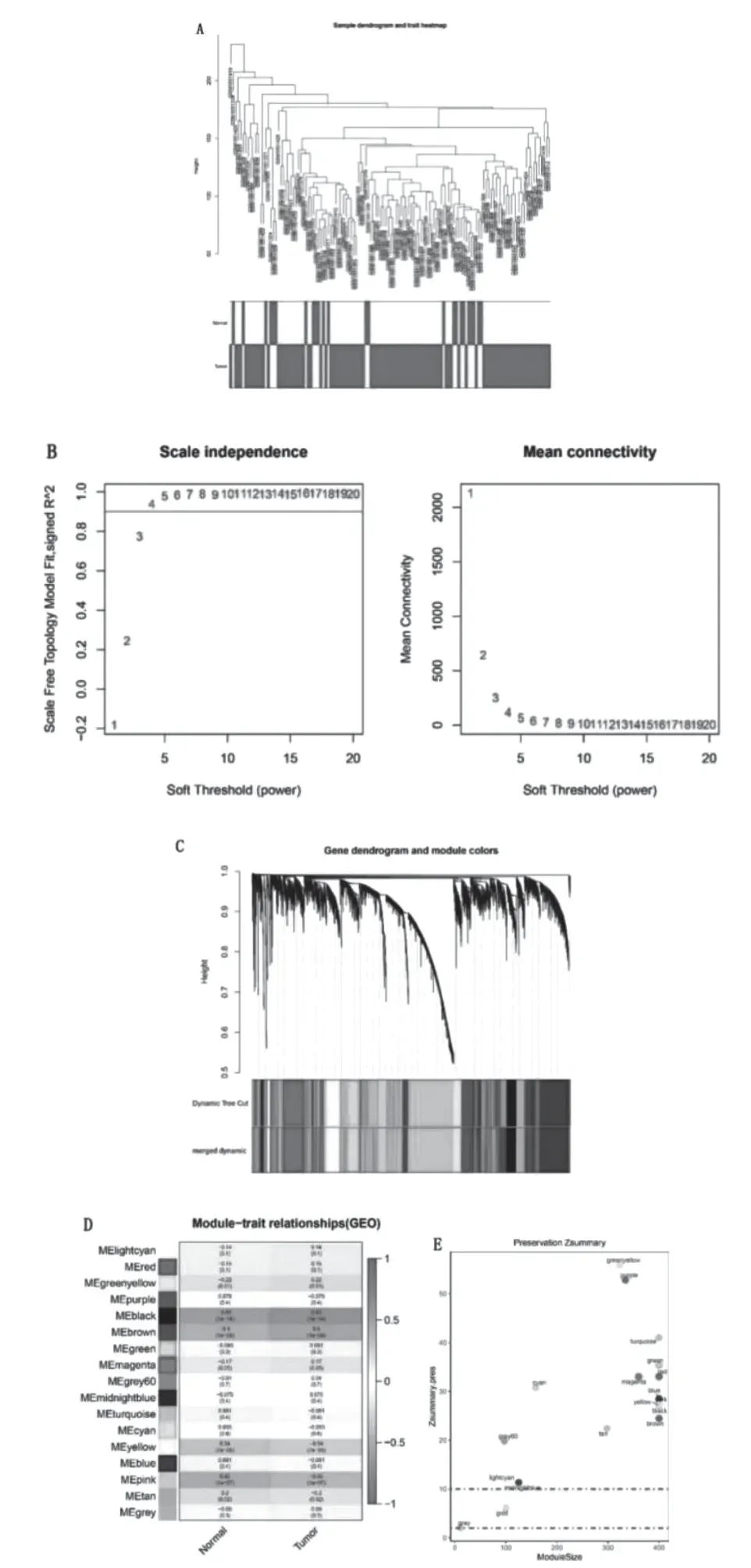
图2 WGCNA共表达分析结果
2.3 蛋白互作网络构建及hub基因筛选
为了从上述125个基因中筛选出最重要的核心基因,我们将这些基因利用String构建蛋白互作网络(图3A),将结果利用Cytoscape进行展示,并使用Cytoscape软件包“CytoHubba”筛选出互作网络中心的10个hub基因(图3B)。这10个基因是蛋白互作网络的核心,它们不仅自身的表达量会在肿瘤细胞中发生显著的变化,还与大多数其他存在表达差异的基因之间具有互作关系。
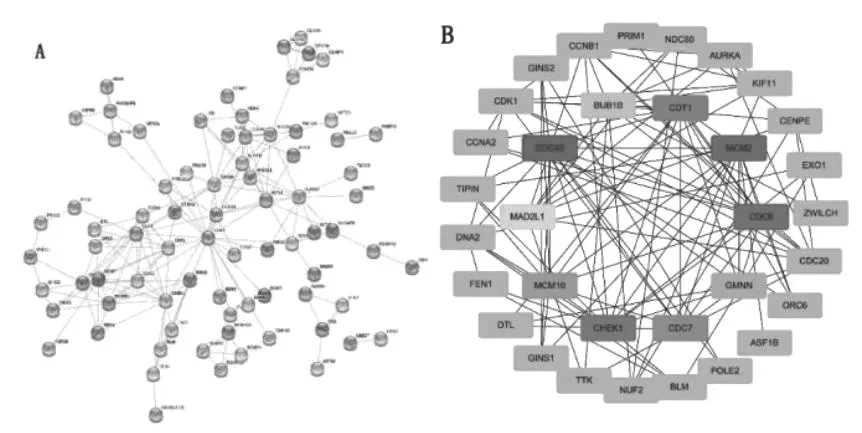
图3 蛋白共表达网络筛选结果
2.4 GO富集分析和KEGG富集分析
为了解差异表达基因的基本表达情况及功能,我们对125个交集基因进行GO富集分析。其中GO富集分析的结果(图4A)显示交集基因主要富集在染色体区域,多参与细胞器分裂,催化微管的结合。KEGG分析的结果(图4B)显示交集基因的作用多富集在细胞周期。该结果提示,这些基因的差异表达可能引起细胞复制分裂的改变,从而引起癌症的发生。

图4 GO富集分析和KEGG富集分析结果
2.5 转录因子共表达网络的构建
为了进一步验证交集基因和癌症的关系,我们将模块基因上传到DAVID后经过分析获得转录因子共表达数据,然后将数据整理后导入到Cytosscape中,修饰过后获得转录因子共表达网络图(图5)。其中E2F和NFY是调控差异基因表达的两个主要的转录因子,图中围绕这两个转录因子的即是受他们调控的基因。底色为蓝色的是在宫颈癌中低表达的基因,底色为红色的是高表达的基因。转录因子E2F是控制细胞周期的关键转录因子之一,它的异常表达会诱导癌症的产生[20]。NFY则与细胞的增殖相关,是癌细胞增殖的标志[21]。
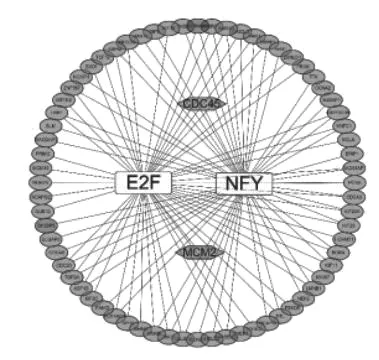
图5 转录因子共表达网络构建结果
2.6 生存分析及无病生存分析
为了验证hub基因与是否在临床上确实影响宫颈癌的预后,我们在GEPIA2上对筛选出的10个hub基因进行生存分析(OS)(图6)。结果显示CDC45和MCM2与患者的生存显著相关。然后利用Oncomine数据库对CDC45和MCM2分析结果进行验证,结果提示CDC45和MCM2的表达和宫颈癌的预后显著相关。
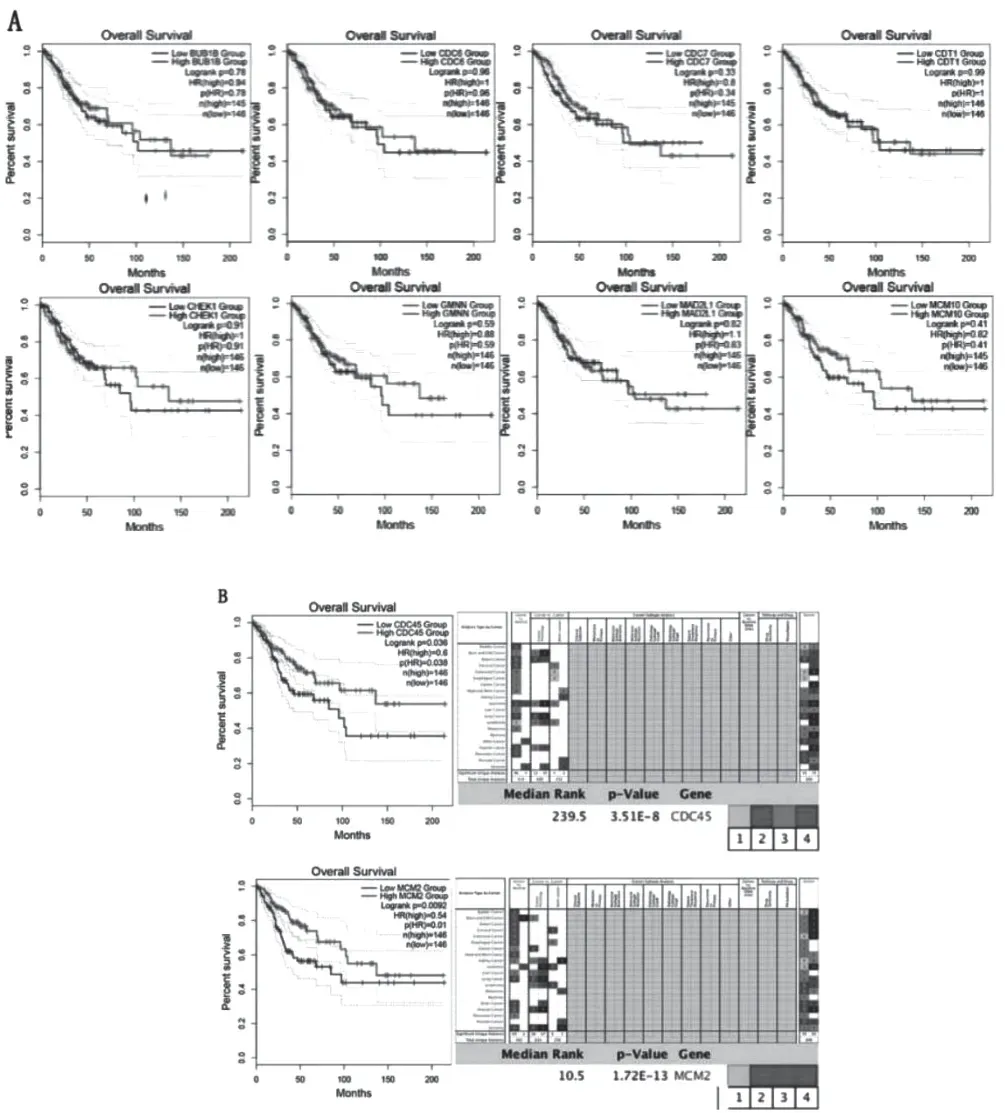
图6 核心基因生存分析结果
2.7 临床相关性分析
为了验证CDC45和MCM2与临床分级之间的关系,我们从GSE63514中提取患者的临床信息,然后用R分析CDC45和MCM2的表达量和患者临床症状之间的相关性(图7)。如图所示为CDC45在宫颈上皮内肿瘤低,中,高三种程度的分化中均有显著差异。MCM2在宫颈上皮内肿瘤的高度分化细胞中,表达量显著高于中、低级分化细胞。这一结果提示我们CDC45和MCM2的表达量和癌细胞的分化程度之间存在关系。
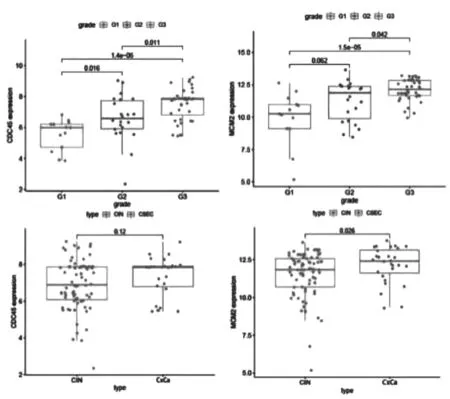
图7 临床性状的相关性分析结果
3 讨论
本研究通过有别于传统差异分析的构建WGCNA共表达网络的方法,筛选出FCDC45、MCM2、CDC6、
CHEK1、CDT1、CDC7、MCM10、GMNN、BUB1B、MAD2L1等10个核心基因。然后通过GO富集分析、KEGG富集分析、转录因子富集分析、生存分析和临床相关分析等多种分析手段,逐步验证了CDC45和MCM2在宫颈癌的发生发展中起着显著的影响。
CDC45是真核DNA15解旋酶的重要组成部分,对DNA的复制有重大影响。它会与单链DNA结合蛋白(replication protein A,RPA)结合并引导RPA和单链DNA结合[22]。而且有研究表明,CDC45是维持延伸过程中复制叉正常运作的重要蛋白。当CDC45上调时会导致细胞凋亡[23
]。根据本研究中GO分析的结果指出,CDC45主要富集在复制叉,在DNA复制的过程中发挥着引导蛋白与DNA结合的作用。KEGG富集分析的结果显示CDC45会富集在细胞周期。转录因子共表达网络的结果显示CDC45会受NFY和E2F的调控影响。其中NFY驱动细胞周期调控基因的转录,是增殖调控的关键角色。高表达与癌症相关[21]。E2F是控制细胞增殖的关键,它和RB口袋蛋白家族、MuvB复合物以及B-MYB和FOXM1调控着与细胞周期相关基因的表达,其异常表达与癌症相关[20],在乳腺癌,前列腺癌,肝癌,消化系统癌症及视网膜母细胞瘤等多种癌症组织中有特异性表达[24-28]。受NFY和E2F所调控的CDC45也与宫颈癌之间存在关联。
本研究中生存分析的结果显示,CDC45的高表达与较长的生存期显著相关,与文献中所描述的CDC45的高表达会诱导细胞更快凋亡的结论相符[21]。临床分析结果显示,CDC45的表达量在不同分化程度癌细胞中均有显著差异,分化程度越低表达量越低。这种现象与生存分析的结果一致。
MCM2是MCM家族中的一员。由MCM2参与所组成的MCM2-7复合体与细胞周期和DNA复制相关,在DNA复制的起点驱动DNA解旋。MCM的磷酸化与DNA复制以及细胞循环进展和检查点响应相关[29]。在癌症方面,有研究表明MCM与细胞周期进程,基因组稳定性相关,其磷酸化异常与癌症的发生和发展相关[30]。在肺癌等共14种癌细胞中MCM2都有显著的高表达,且MCM2已经作为一种癌症标志物被用于判断病情和患者的预后[31]。
根据本研究中GO富集分析的结果显示,MCM2在细胞周期中有DNA催化活性。KEGG富集分析显示MCM2参与了细胞周期和DNA复制过程。转录因子的富集分析结果显示,MCM2的表达受E2F的调控影响。这也表示MCM2与癌症的产生发展相关。
本研究生存分析结果显示,在宫颈癌中,MCM2的高表达会显著提高患者的预后效果,这一结果与其他种类的癌症研究的结果不符。究其原因可能是与MCM2的磷酸化会抑制DNA合成[30]。临床相关性分析则指出,MCM2在高分化程度的细胞中的表达量显著高于中低分化程度的细胞,该结果与生存分析的结果一致。
此外已有研究提出,MCM2和CDC45都是CMG复合体的组成部分,该复合体具有DNA解旋酶功能。CDC45在细胞核中参与DNA的复制[32],细胞则会通过60merssDNA与CDC45相互作用来阻止复制[33],而CDC45与MCM2和MCM5的是CMG复合物构成的关键[34]。由此我们可以推断,在宫颈癌中CDC45和MCM2之间的相互作用也会对病情产生影响,其作用机制和作用效果则有待后续实验研究。
综上所述,CDC45和MCM2在宫颈癌中的作用及机制有进一步研究挖掘的价值,CDC45和MCM2的表达量在临床上对于宫颈癌这一疾病的产生、发展状态的诊断以及预后结果的推测也均有一定的参考价值。
猜你喜欢
好日子(2021年8期)2021-11-04
海峡姐妹(2020年2期)2020-03-03
保健与生活(2019年3期)2019-08-01
华人时刊(2018年17期)2018-11-19
海峡姐妹(2018年7期)2018-07-27
数学学习与研究(2018年7期)2018-05-16
山东青年(2017年11期)2018-03-29
瞭望东方周刊(2017年36期)2017-09-28
都市丽人(2015年4期)2015-03-20
中国民族民间医药·下半月(2014年4期)2014-09-26
