
融合蛋白GST-Ulp1p在大肠杆菌中的高效可溶性表达及其活性鉴定
2010-10-16 08:08傅俊华王琪尹杰超刘铭瑶李宁姚文斌任桂萍李璐李德山
生物工程学报 2010年6期
傅俊华,王琪,尹杰超,刘铭瑶,李宁,姚文斌,任桂萍,李璐,李德山
东北农业大学生命科学学院 生物制药实验室,哈尔滨 150030
融合蛋白GST-Ulp1p在大肠杆菌中的高效可溶性表达及其活性鉴定
傅俊华,王琪,尹杰超,刘铭瑶,李宁,姚文斌,任桂萍,李璐,李德山
东北农业大学生命科学学院 生物制药实验室,哈尔滨 150030
利用基因工程技术,体外重组小分子类泛素修饰蛋白酶1(Ulp1)的活性片段,获得高表达、高特异性重组蛋白酶。从酿酒酵母Saccharomyces cerevisia中提取Ulp1编码第403到621个氨基酸残基之间的DNA片段(Ulp1p),在其C端加入6×His并连接到大肠杆菌表达载体pGEX中,构建重组表达质粒pGEX-Ulp1p-his6。将重组质粒转化至大肠杆菌Rosetta(DE3)中,氨苄青霉素抗性筛选转化子。表达、纯化后,以SUMO融合蛋白检测其活性。经过优化,该蛋白可溶性表达,表达量占菌体总蛋白的40.12%。可通过谷胱甘肽琼脂糖凝胶柱或Ni-NTA凝胶亲和层析纯化得到纯度98%的蛋白。经酶切分析,比活力为1.375×104U/mg。融合蛋白GST-Ulp1p-His6无需切除谷胱甘肽S-转移酶(GST)标签,具有很高的活性,制备简易;6×His标签,有利于底物蛋白切割后纯化,减少蛋白损失。本研究为制备高活力的SUMO蛋白酶提供了一个新方法。
SUMO蛋白酶1(Ulp1),谷胱甘肽S-转移酶(GST),高效表达,活性鉴定
Abstract:The aim of the study is to obtain an efficient expression of recombinant ubiquitin-like specific protease 1(Ulp1)by gene engineering.We cloned the Ulp1p, active fragment(403 aa−621 aa)of Ulp1, fromSaccharomyces cerevisia, and subcloned into pGEX/Rosetta(DE3)to form an expression plasmid, pGEX-Ulp1p-His6.In order to enhance the solubility of GST-Ulp1p-His6,we purified the fusion protein GST-Ulp1p-His6by either glutathioneS-transferase agarose or Ni-NTA resin chromatography, the purity was up to 98%.We utilized the protein to cleave the SUMO fusions, and the specific activity of GST-Ulp1p-His6was 1.375×104U/mg.This study showed that the recombinant protein GST-Ulp1p-His6displayed high specificity and activity.
Keywords:ubiquitin-like specific protease 1(Ulp1), glutathioneS-transferase(GST), high-level expression, activity identification
小分子类泛素修饰蛋白(Small ubiquitin-like modifier protein,SUMO)广泛存在于各种真核细胞中,参与调节细胞凋亡、信号转导、RNA转录、蛋白的核质运输以及细胞周期等多种生理进程[1]。由SUMO介导的蛋白质翻译后修饰是对其所修饰的蛋白质功能与定位的一个关键性调节机制。近年来,SUMO被发现可以作为重组蛋白表达的融合标签和分子伴侣,具有抗蛋白酶水解、显著增加重组蛋白表达量以及促进靶蛋白正确折叠、提高可溶性等功能[2-3]。
SUMO在真核生物中的修饰作用是可逆的,经SUMO修饰的蛋白可以通过SUMO蛋白酶切割成底物蛋白和 SUMO,这个过程为去 SUMO化(Desumoylation)[4]。至今已鉴定了 7个 SUMO蛋白酶家族成员,其定位不同,功能略有差异,具有不同的底物特异性[5]。SUMO蛋白酶在真核生物体内主要行使2种生理功能:1)切除SUMO前体C-末端的几个氨基酸残基,以暴露C-末端的Gly-Gly残基使之成为具修饰、转运等功能的成熟 SUMO;2)将SUMO与目的蛋白偶合物水解成SUMO和目的蛋白。研究资料显示,各种SUMO蛋白酶都含有一个保守的 C-末端 ULP结构域(约 200个氨基酸),该结构域具有催化活性[6-7];不同的SUMO蛋白酶有不同的N-末端结构域,正是这种差异将不同的SUMO蛋白酶定位于细胞的不同部分[8-10]。在所有 SUMO蛋白酶中,研究得最详细的是酿酒酵母中的蛋白酶1——Ulp1。它是由621个氨基酸残基组成的蛋白质多肽,至少含有2个结构域,一个是保守的C-端蛋白酶折叠区域(432~621 aa);另一个是保守性弱的N-端结构域(1~432 aa)。通过序列相似性对比、结构预测和选择性的蛋白水解实验发现,Ulp1中第403~621 aa活性片段Ulp1p表现出具有全长的Ulp1酶切活性,并能够水解以α-氨基连接的SUMO-目的蛋白偶合物。利用Ulp1与SUMO相互作用的晶体结构发现Ulp1对SUMO的识别主要依据底物的三级结构,而不像其他蛋白酶只识别特定的氨基酸序列;Ulp1的水解活性对SUMO-蛋白偶合物具有高效性和特异性,这些独特的优点使得 Ulp1可以作为一种新的工具酶用于重组蛋白的切割和纯化。
本实验采用pGEX表达蛋白,其优越性之一是载体构建无需考虑SD及RBS序列对基因的转录和翻译效率的影响,多克隆位点在GST基因之后,无论外源基因以何种接口插入,都不会改变这个载体的SD及RBS序列,从而表达出GST融合蛋白。其表达量虽高,但可溶性目的蛋白所占比例较小。本实验通过对表达条件的一系列优化,最终得到高表达、可溶的目的蛋白。本实验在引物中设计多聚组氨酸标签连在目的蛋白C-末端,因此在进一步的纯化中,既可利用谷胱甘肽琼脂糖凝胶柱纯化,也可通过 Ni-NTA凝胶亲和层析得到纯化蛋白。本研究为高效表达特异性重组可溶的SUMO蛋白酶提供了一条新的途径。
1 材料和方法
1.1 材料
1.1.1 菌株、质粒及主要试剂
酿酒酵母菌株购自黑龙江省微生物研究所;E.coliRosetta(DE3)由本实验室保存;pGEX-6P-1购自Amersham Pharmacia Biotech;PMD18 T-simple-Vector购自TaKaRa;Ni-NTA Agarose购自Qiagen;蛋白分子量标准购自 Fermentas;兔抗酵母 Ulp1多抗购自 LifeSpan BioSciences;活性测定底物蛋白SUMO-IL-1β、SUMO-TNFα和 SUMO-FGF-21由本实验室提供。
引物:P1:F 5′-CGCGGATCC ATGCTTGTTCCT GAATTAAATGA-A-3′,下划线部分为BamH I酶切位点。P2:R 5′-CCGCTCGAG CTAGTGATGATGATGA TGATGTT-TTAAAGCGTCGGTT-3′,下划线部分依次为XhoI酶切位点和多聚组氨酸标签。
1.2 方法
1.2.1 Ulp1p基因的克隆及重组表达载体的构建
提取酿酒酵母基因组DNA,利用引物P1和P2进行PCR扩增,扩增产物经回收纯化后利用BamH I和XhoI对其进行酶切。此酶切产物再次经胶回收与经BamH I和XhoI酶切后的pGEX-6P-1载体连接,构建重组质粒pGEX-Ulp1p-His6,此质粒经测序正确后转化E.coliRosetta(DE3)。
1.2.2 目的蛋白的诱导表达
将转入重组质粒 pGEX-6P-1-Ulp1p-His6的Rosetta(DE3)划线培养并挑取单菌落接种于LB培养液中(含100 mg/L氨苄青霉素),振荡培养过夜。将上述培养物按1%接种于LB培养液中(含100 mg/L氨苄青霉素),培养至OD600值为0.3~0.4时加入IPTG进行诱导表达。取不同诱导温度、诱导转速、诱导时间及不同IPTG浓度的诱导菌经超声破碎后离心,将沉淀部分加入与上清相同体积的蒸馏水,取菌体上清部分及沉淀部分进行 SDS-PAGE,检测融合蛋白的表达及可溶性情况。
1.2.3 融合蛋白GST-Ulp1p-His6的纯化
取pGEX-Ulp1p-His6转化菌大量诱导表达。诱导后的菌体用裂解缓冲液(10 mmol/L imidazole,300 mmol/L NaCl,50 mmol/L NaH2PO4,pH 8.0)重悬后加入溶菌酶至终浓度为 1 mg/mL,冰上放置30 min后超声破碎,10 000×g、4℃离心30 min,收集上清。上清利用 AKTA-purifier100系统经过HisTrapTM FF crude亲和层析,用清洗缓冲液(20 mmol/L imidazole,300 mmol/L NaCl,50 mmol/L NaH2PO4,pH 8.0)洗去杂蛋白后,再用洗脱缓冲夜(250 mmol/L imidazole,300 mmol/L NaCl,50 mmol/L NaH2PO4,pH 8.0)洗脱,收集唯一的洗脱峰即为融合蛋白。融合蛋白含有GST标签,因此,也可通过谷胱甘肽琼脂糖凝胶柱进行纯化,将细菌用超声波破壁后离心的上清液与50%谷胱甘肽琼脂糖凝胶珠混合,15~30 min后,用过量的游离谷胱甘肽竞争洗脱。结合缓冲液(140 mmol/L NaCl,2.7 mmol/L KCl,10 mmol/L Na2HPO4,1.8 mmol/L KH2PO4,pH 7.3),洗脱缓冲夜(50 mmol/L Tris-HCl,10 mmol/L还原型谷胱甘肽,pH 8.0)。收集的融合蛋白脱盐后进行SDS-PAGE 电泳检测。
1.2.4 融合蛋白GST-Ulp1p-His6的Western blotting检测
取纯化后的融合蛋白经SDS-PAGE电泳后,将蛋白转移至NC膜上,50 g/L脱脂奶粉室温封闭1 h,加入兔抗酵母Ulp1多抗,4℃孵育过夜。PBST洗膜3次,每次5 min。再加入HRP标记的羊抗兔IgG,37℃温育1 h,PBST和PBS各洗膜3次后,ECL化学发光显影。
1.2.5 融合蛋白GST-Ulp1p-His6的活性检测
以SUMO-TNFα、SUMO-FGF-21和SUMO-IL-1β作为底物蛋白,加入GST-Ulp1p-His6和终浓度为2 mmol/L的 DTT,30℃切割 1 h,产物进行 SDSPAGE电泳检测。
1.2.6 比活力的初步测定
以底物蛋白SUMO-IL-1β为例,GST-Ulp1p-His6分别以 13 µg/mL、26 µg/mL、52 µg/mL 和 78 µg/mL的终浓度对底物进行切割,2 mmol/L的DTT,30℃切割1 h,产物进行SDS-PAGE电泳检测。以30℃下反应1 h,切割>85%的2 µg对照底物所需要的酶量定义为一个活性单位。根据切割情况,得出酶的活力单位及比活力。
2 结果
2.1 重组质粒的构建和鉴定
利用引物P1和P2克隆产物经琼脂糖电泳检测,发现获得位于500 bp和750 bp之间的单一条带,与预期的699 bp(Ulp1p基因与酶切位点核苷酸之和)大小相符。将重组质粒 pGEX-Ulp1p-His6测序,测序结果与 GenBank上的序列(Accession No.Q02724)完全一致,证实Ulp1p基因已成功克隆并正确连接到表达载体中(图略)。
2.2 融合蛋白表达条件的优化
在37℃ 培养条件下0.25 mmol/L IPTG诱导4 h,利用pGEX-6P-1表达GST-Ulp1p-His6融合蛋白时,在分子量约53 kDa处出现一条新增蛋白条带,与预期结果相符,融合蛋白主要以包涵体形式存在(图1),选取0.25 mmol/L IPTG诱导表达,取不同诱导温度(37℃,20℃)、不同转速(100 r/min,55 r/min)、不同诱导时间(1~12 h)的诱导菌进行18% SDS-PAGE,结果显示目的蛋白的表达量达到高峰时的参数为:20℃,55 r/min,11 h。IPTG浓度为0.05 mmol/L时,上清所占比率最高。经灰度扫描分析,表达的融合蛋白约占菌体总蛋白的40.12%(图2)。
2.3 融合蛋白 GST-Ulp1p-His6的纯化及 Western blotting鉴定
利用pGEX-6P-1载体表达外源蛋白时,融合蛋白N端含有GST标签,可以利用谷胱甘肽琼脂糖凝胶柱进行纯化;本实验在目的蛋白 C-末端设计有6×His标签,也可以利用Ni-NTA层析柱进行纯化。表达菌破碎后上清经亲和层析得到唯一的洗脱峰即为 GST-Ulp1p-His6融合蛋白。蛋白样品经 18%SDS-PAGE电泳显示,蛋白分子量约为53 kDa,灰度扫描结果显示纯化后成熟 GST-Ulp1p-His6纯度达98%(图3)。Western blotting分析表明,该融合蛋白可与兔抗酵母Ulp1的单抗发生特异性反应,说明表达的是SUMO蛋白酶1活性片段(图3)。
2.4 融合蛋白GST-Ulp1p-his6的活性检测
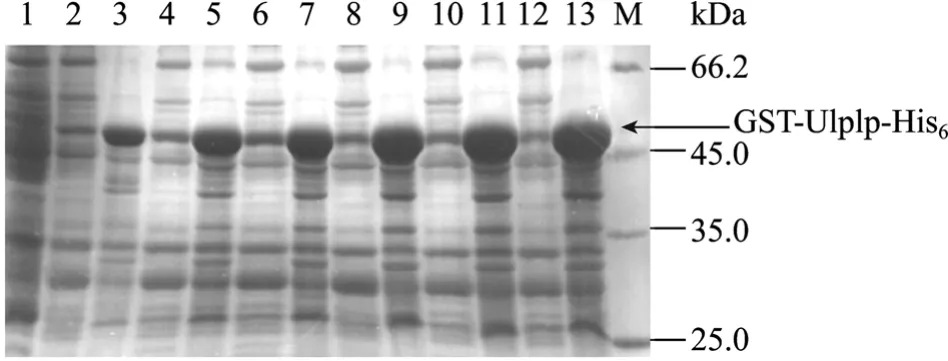
图1 融合蛋白GST-Ulp1p-His6的SDS-PAGE电泳分析Fig.1 SDS-PAGE analysis of the fusion protein GST-Ulp1p-His6.1: uninduced cell lysate; 2, 4, 6, 8, 10, 12: supernatant of cell lysate induced by 0.25 mmol/L IPTG at 37°C for 1,2, 3, 4, 5, 6 h; 3, 5, 7, 9, 11: inclusion body induced by 0.25 mmol/L IPTG at 37°C for 1, 2, 3, 4, 5, 6 h; M: protein molecular weight marker.
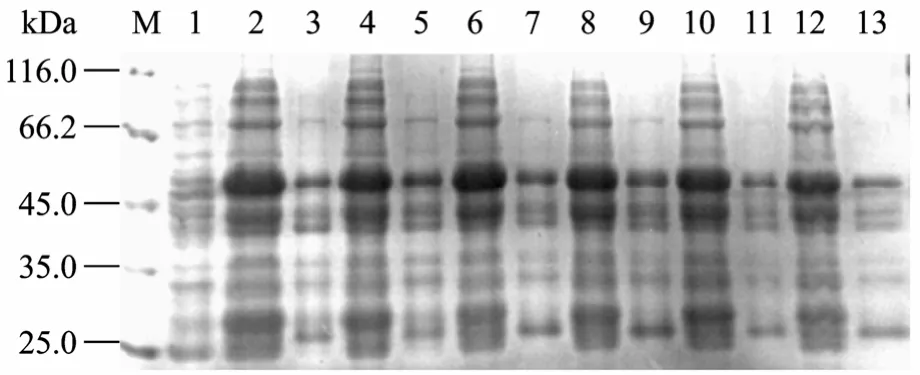
图2 融合蛋白GST-Ulp1p-His6的SDS-PAGE电泳分析Fig.2 SDS-PAGE analysis of the fusion protein GST-Ulp1p-His6.M: protein molecular weight marker; 1: uninduced cell lysate; 2, 4, 6, 8, 10, 12: supernatant of cell lysate induced by 0.05, 0.1, 0.2, 0.5, 1, 2 mmol/L IPTG at 20°C for 11 h; 3, 5, 7,9, 11, 13: inclusion body induced by 0.05, 0.1, 0.2, 0.5, 1,2 mmol/L IPTG at 20°C for 11 h.
以SUMO-TNFα、SUMO-FGF-21和SUMO-IL-1β作为底物蛋白,加入GST-Ulp1p-His6和终浓度为2 mmol/L的 DTT,30℃切割 1 h。产物经 18%SDS-PAGE电泳分析,结果显示,SUMO-TNFα、SUMO-FGF-21和 SUMO-IL-1β能够被 GST-Ulp1p-His6完全切割(图4)。
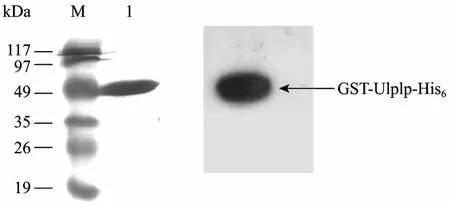
图3 纯化后融合蛋白的SDS-PAGE电泳分析及Western blotting鉴定Fig.3 SDS-PAGE analysis of the purification and western blotting of GST-Ulp1p-His6.M: protein molecular weight marker; 1: purified GST-Ulp1p-His6.
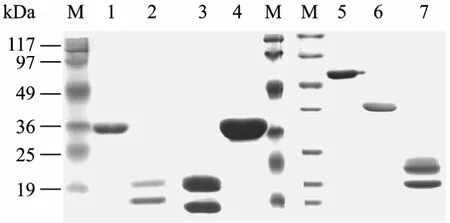
图4 GST-Ulp1p-His6切割SUMO融合蛋白的SDS-PAGE电泳分析Fig.4 SDS-PAGE analysis of the cleavage of GST-Ulp1p-His6on SUMO-TNFα, SUMO-FGF-21 and SUMO-IL-1β.M: protein molecular weight marker; 1: SUMO-TNFα; 2: cleavage products of SUMO-TNFα; 3: cleavage products of SUMO-FGF-21; 4:SUMO-FGF-21; 5: GST-Ulp1p-His6; 6: SUMO-IL-1β; 7: cleavage products of SUMO-IL-1β.
2.5 比活力的初步测定
以 SUMO-IL-1β为底物蛋白,GST-Ulp1p-His6分别以 13、26、52、78 µg/mL的终浓度对 110 µg底物进行切割,2 mmol/L DTT,在150 µL酶切缓冲液中30℃反应1 h,产物进行18% SDS-PAGE电泳检测。结果显示,分子量为37 kDa的SUMO-IL-1β被切割为 20 kDa的 SUMO和 17 kDa的 IL-1β。26 µg/mL的酶切割>85%的底物蛋白。根据酶活力定义,酶活力单位为5.5 U/µL,比活力为1.375×104U/mg(图5)。
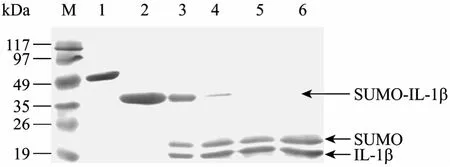
图5 不同酶浓度切割SUMO-IL-1β的SDS-PAGE电泳分析Fig.5 SDS-PAGE analysis of the cleavage of different concentration of GST-Ulp1p-his6on SUMO-IL-1β.M: protein molecular weight marker; 1: GST-Ulp1p-His6; 2: SUMO-IL-1β;3−6: concentrations of GST-Ulp1p-His6were 13, 26, 52,78 µg/mL for cleavage of SUMO-IL-1β.
3 讨论
温度对于融合蛋白 GST-Ulp1p-His6的诱导表达有一定的影响。在实验过程中,37℃培养条件下,融合蛋白全部以包涵体形式存在。随着培养温度的下降,其可溶性表达的情况则越来越好,但是表达量会有所下降,20℃诱导表达时,融合蛋白均为上清表达,表达量可以达到菌体总上清蛋白的 40.12%。IPTG的浓度也影响表达量及可溶情况,从本实验看,IPTG浓度越小,表达量反而越高,上清所占比例也越大,可能是IPTG的毒性杀死细胞,同时也会增加外源蛋白表达速度,容易造成包涵体的形成。该酶含有GST标签,可以通过谷胱甘肽琼脂糖凝胶柱进行纯化,蛋白纯化条件温和,有利于保持蛋白活性。许多用基因工程菌株生产的外源蛋白由于在提取和纯化过程中加入变性剂而改变了蛋白质的空间构象,因而即使保持了正确的一级结构,也不能表现出生物活性或活性很低。而GST融合蛋白,可以直接从细菌裂解液中用固定化谷胱甘肽将它吸附,洗脱条件温和,整个过程无变性剂的加入,最大限度地保持了蛋白的天然构象。在GST融合蛋白的原核表达系统中(例如pGEX系列),GST融合蛋白经常处于不可溶状态。本实验采用低温慢速,低IPTG浓度过夜诱导,使得上清比例达到90%,几乎全部为可溶性蛋白。同时,该酶还设计有多聚组氨酸标签,通过Ni-NTA凝胶亲和层析得到纯化蛋白。通过两种方式获得的蛋白纯度均高达98%,无明显差别,可根据实验条件自行选择。酶切产物在经过Ni-NTA凝胶亲和层析以去掉SUMO和蛋白酶时,由于带有His标签,故可与SUMO同时被洗脱下来,从而节省了洗脱步骤,避免了长时间反复操作而影响蛋白活性。
该融合蛋白酶具有GST标签,可增加其表达量。本研究发现,在酶切反应中,融合的GST-SUMO蛋白酶即具有酶切活性,且活性很高,这在目前的研究中尚属罕见。如切掉GST标签,则需要凝血酶或凝血因子在特异性位点上切除,而该凝血酶价格昂贵,不利于节约成本;且再次切割会影响蛋白收率及活性。本研究无需切掉GST标签,便具有酶切活性,开辟了制备高活力蛋白酶的新途径。
SUMO蛋白酶能够识别完整的SUMO标签蛋白序列,并能高效地把SUMO从融合蛋白上切割下来。由于其识别序列较长,所以SUMO蛋白酶酶切反应有很高的特异性,而且切割后得到的目的蛋白不含任何多余氨基酸,不会影响目的蛋白的活性,这对于那些N末端对活性影响较大的蛋白质的表达是极其重要的。同时SUMO蛋白酶进行切割时识别的是底物的三级结构,避免了由于切割传统融合标签的蛋白酶识别的氨基酸序列一般较短而可能在目的蛋白内部进行的错误切割[11]。
利用SUMO融合表达系统表达重组蛋白,在分离靶蛋白过程中,必须使用SUMO蛋白酶作为切割工具。本实验成功构建并高效表达了可溶性融合蛋白GST-Ulp1p-His6,并可以切割各种SUMO融合蛋白,在本实验室得到广泛应用。由于SUMO蛋白酶市场价格仍然昂贵,因此,大量表达高活力和高特异性的重组SUMO蛋白酶具有十分重要的经济意义和市场价值。
REFERENCES
[1]Kerscher O.SUMO junction-what's your function? New insights through SUMO-interacting motifs.EMBO Rep,2007, 8(6): 550–555.
[2]Butt TR, Edavettal SC, Hall JP,et al.SUMO fusion technology for difficult-to-express proteins.Protein Expr Purif, 2005, 43(1): 1–9.
[3]Su ZJ, Huang YD, Zhou QN,et al.High-level expression and purification of human epidermal growth factor withSUMO fusion inEscherichia coli.Protein Pept Lett, 2006,13(8): 785–792.
[4]Ivan M, Kondo K, Yang H,et al.HIF(alpha)targeted for VHL-mediated destruction by proline hydroxylation:implications for O(2)sensing.Science, 2001, 292(5516):464–468.
[5]Johnson ES.Protein modification by SUMO.Annu Rev Biochem, 2004, 73: 355–382.
[6]Li SJ, Hochstrasser M.A new protease required for cell-cycle progression in yeast.Nature, 1999, 398(6724):246–251.
[7]Mossessova E, Lima CD.Ulp1-SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in yeast.Mol Cell, 2000, 5(5): 865–876.
[8]Li SJ, Hochstrasser M.The Ulp1 SUMO isopeptidase:distinct domains required for viability, nuclear envelope localization, and substrate specificity.J Cell Biol, 2003,160(7): 1069–1081.
[9]Panse VG, Kuster B, Gerstberger T,et al.Unconventional tethering of Ulp1 to the transport channel of the nuclear pore complex by karyopherins.Nat Cell Biol, 2003, 5(1):21–27.
[10]Hang J, Dasso M.Association of the human SUMO-1 protease SENP2 with the nuclear pore.J Biol Chem, 2002,277(22): 19961–19966.
[11]Marblestone JG, Edavettal SC, Lim Y,et al.Comparison of SUMO fusion technology with traditional gene fusion systems: enhanced expression and solubility with SUMO.Protein Sci, 2006, 15(1): 182–189.
Expression and characterization of soluble recombinant Ulp1p with glutathione S-transferase tag in Escherichia coli
Junhua Fu, Qi Wang, Jiechao Yin, Mingyao Liu, Ning Li, Wenbin Yao, Guiping Ren, Lu Li,and Deshan Li
Biopharmaceutical Laboratory, College of Life Science, Northeast Agricultural University, Harbin 150030, China
Received:January 8, 2010;Accepted:March 17, 2010
Supported by:National Natural Science Foundation of China(No.30700591).
Corresponding author:Deshan Li.Tel: +86-451-55190645; E-mail: Deshanli@163.com国家自然科学基金(No.30700591)资助。
猜你喜欢
云南化工(2021年6期)2021-12-21
太原理工大学学报(2021年6期)2021-11-25
中国医学影像学杂志(2021年6期)2021-08-13
科学(2020年2期)2020-08-24
中国海洋大学学报(自然科学版)(2019年7期)2019-05-21
中国海洋大学学报(自然科学版)(2019年7期)2019-01-04
广州化工(2016年11期)2016-09-02
天然产物研究与开发(2016年6期)2016-06-05
中国继续医学教育(2015年3期)2016-01-06
生物技术通报(2015年1期)2015-04-10
