
吡啶甲酸铑阳离子催化甲醇羰基化反应IRC解析及催化剂结构与性能的理论计算
2011-01-06 07:52吉文欣刘翔宇冀永强
湖北大学学报(自然科学版) 2011年4期
吉文欣,刘翔宇,冀永强
(宁夏大学宁夏能源化工重点实验室,宁夏 银川750021)
吡啶甲酸铑阳离子催化甲醇羰基化反应IRC解析及催化剂结构与性能的理论计算
吉文欣,刘翔宇,冀永强
(宁夏大学宁夏能源化工重点实验室,宁夏 银川750021)
分别采用从头算HF方法和密度泛函(DFT)B3LYP方法,在LANL2DZ基组下,应用内禀反应坐标(IRC)理论分析吡啶甲酸铑阳离子催化甲醇羰基化决速步骤反应沿最小能量途径(MEP)上相互作用分子间化学键的变化,对比[Rh(CO)2I2)]-催化剂,从催化剂结构、分子轨道分布等方面,探讨吡啶甲酸铑阳离子催化剂具有较高活性的本质原因;同时也从分子轨道和构型分析上总结吡啶甲酸铑阳离子催化剂的缺陷,刚性的五元环结构和离域π键的存在不利于协同决速步骤反应的结构挠变,并提出优化催化剂构型的有效途径.
HF方法;密度泛函DEF;铑;催化剂;构型
乙酸是一种应用广泛的重要化工原料,主要被用于合成乙酸乙烯酯的单体(VAM)、合成乙酸酐的原料以及生产精制对苯二甲酸(PTA)的溶剂等.乙酸的合成方法主要有碳水化合物的发酵,石脑油或正丁烷的氧化,乙烯或乙醛的氧化以及甲醇的羰基化等方法[1-2].其中,甲醇羰基化法占目前世界乙酸生产量的60%.1968年美国Monsanto公司的Paulik和Roth报道的羰基铑碘化合物([Rh(CO)2I2)]-)催化剂体系[3]对甲醇羰基化合成乙酸具有很高的催化活性和选择性,而且反应条件十分温和,从而被冠以“低压法”之美名.但是,铑碘催化剂易沉淀失活,近年来针对这一缺点所进行的改进一直是研究热点[4-7],其中采用吡啶甲酸类配体,利用其中的共轭氮和羰基氧与铑形成含有两种不同配位键(N→Rh和O→Rh)的螯合型吡啶甲酸铑阳离子配合物([MRh(CO)2]+,M:吡啶甲酸配体)[8-10],对催化甲醇羰基化合成乙酸具有很高的活性和稳定性.
在前期工作中,我们在理论上研究了该催化剂的反应机理,CH3OH与CO在吡啶甲酸铑阳离子催化剂的作用下反应分4步进行,分别是碘甲烷氧化加成反应、羰基重排反应、羰基配位反应、CH3COI还原消除基元反应,其中碘甲烷氧化加成反应(如图1)位垒最高(120.91kJ/mol),是整个反应的决速步骤[11].然而仅仅知道这一点,从理论上指导更高效催化剂的合成,从而达到催化剂分子设计的目的还远远不够.在前期工作的结论之下,考虑到HF方法忽略了大部分的电子相关,必然带来较大的能量误差,我们采用DFT方法B3LYP泛函[12];同时由于催化剂含有一个比较重的过渡金属Rh,此时相对论效应的影响已经不可忽视,所以本文中采用包含了重金属相对论效应修正的赝势基组LANL2DZ,应用IRC理论分析了决速步骤反应沿最小能量途径(MEP)上相互作用分子间化学键的变化,对比[Rh(CO)2I2)]-催化剂[13-15],从催化剂结构、电荷分布、分子轨道分布等方面,深入探讨了吡啶甲酸铑阳离子催化剂具有较高活性的本质原因,并依据计算结果探讨了[MRh(CO)2]+催化剂的缺陷,并提出了催化剂构型优化的方向.
1 研究方法
采用从头算 HF方法和密度泛函(DFT)B3LYP方法,采用赝势基组(LANL2DZ),优化了[Rh(CO)2I2)]-和[MRh(CO)2]+催化剂以及它们催化甲醇羰基化决速步骤反应(碘甲烷氧化加成)反应物、产物、过渡态和中间体的构型,应用IRC理论分析了该反应沿最小能量途径(MEP)上相互作用分子间化学键的变化,对每一驻点作了零点能(zero-point energy,ZPE)校正,计算了反应位垒,分析了催化剂构型、分子轨道能级分布与活性之间的关系.所有计算采用Gaussain 03量子化学计算软件完成[16].
2 结果与讨论
2.1 碘甲烷氧化加成反应各驻点构型及IRC解析碘甲烷氧化加成是整个催化过程的决速步骤,该步反应并不是一步就完成的,它经历了一次构型的调整.反应物之一的[MRh(CO)2]+(Cat1)在稳态时,中心原子Rh15与它的4个配位原子N10、O12、C16和C17基本在同一平面上,形成一个以Rh为中心的四配位体正方平面结构,反应开始时,CH3I沿着与催化剂平面垂直的方向向金属中心Rh接近,在接近的过程中,原有的四配位正方平面结构逐渐被破坏,首先是I与[MRh(CO)2]+的Rh配位,经过过渡态TS1,生成一个较稳定的中间体(INT),然后经过过渡态TS2,H3C—I键逐渐断开,CH3迁移从I上迁移到Rh上.整个加成反应经历了两个过渡态,经过一个稳定中间体,催化剂构型得以调整,使得CH3I氧化加成反应总体位垒降低,[MRh(CO)2]+的活性高于[Rh(CO)2I2)]-.从反应各驻点的几何结构(图1)能够发现,先是I的加成,然后才是CH3的迁移,这中间二面角C16-Rh15-O12-C11一直在变化(180.0°~14.2°),实际上是 Rh15-N10-O12平面与 Rh15-C16-C17平面之间由基本在同一平面到基本垂直最后又回到同一平面的过程,CH3I加成后两个CO交换位置,生成加成产物Cat2,此时Rh由四配位正方平面结构变为六配位四面双锥结构.
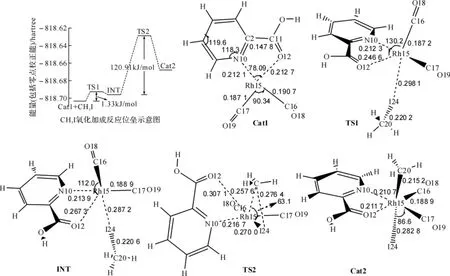
图1 CH3I氧化加成反应过程中位垒示意图和各驻点几何结构(键长:nm,键角:°)
应用IRC理论,我们分析了主要反应原子间距离沿MEP的变化(图2)可以看出,第一步反应,Cat1-TS1-INT的过程(图2(a))主要的键长变化发生在Rh-O12、Rh-I24之间,其他化学键变化很小,随反应进行Rh-O12不断拉长,趋于断开,Rh-I24不断缩短,趋于成键.第二步反应,INT-TS2-Cat2的过程(图2(b))主要的键长变化发生在Rh-C20之间,其实这一步反应是—CH3从I到Rh的转移加成.从整个CH3I氧化加成反应来看,可以看出Rh-O12键长的变化贯穿两步反应,Rh-O12键由不断拉长到基本断开,然后又恢复,直至生成Cat2.CH3I氧化加成反应伴随Rh-O12键的开合以及Rh15-N10-O12平面与Rh15-C16-C17平面之间相对位置的调整,这是催化剂反应过程中的一种结构上的挠变,这种挠变在反应中会增加反应位阻,使得过渡态位能升高,最终导致反应位垒升高,限制了催化活性的增加.

图2 CH3I氧化加成反应主要原子间距离沿MEP的变化
2.2 碘甲烷氧化加成反应各驻点能量及反应位垒表1是[Rh(CO)2I2)]-和[MRh(CO)2]+催化剂碘甲烷氧化加成反应中各驻点的能量(包含零点能校正能),可以看出采用B3LYP方法得到的能量都低于HF方法得到的能量,这是由于HF方法忽略了大部分的电子相关,必然带来较大的能量误差,而B3LYP方法由于比较好地估计了电子相关能,得到的能量更低,尤其在计算包含过渡金属的化合物时结果相对准确,得到的反应位垒也相对准确.但是不论是哪种方法都反映出在决速步骤CH3I氧化加成反应中,[MRh(CO)2]+催化剂比[Rh(CO)2I2)]-催化剂具有更低的反应位垒,催化活性更强.[Rh(CO)2I2)]-(Cat1)在CH3I氧化加成时没有中间体生成,一步生成了CH3I加成产物(Cat2),这也导致该催化剂在CH3I氧化加成反应中位垒较高.这与实验结果一致[8].
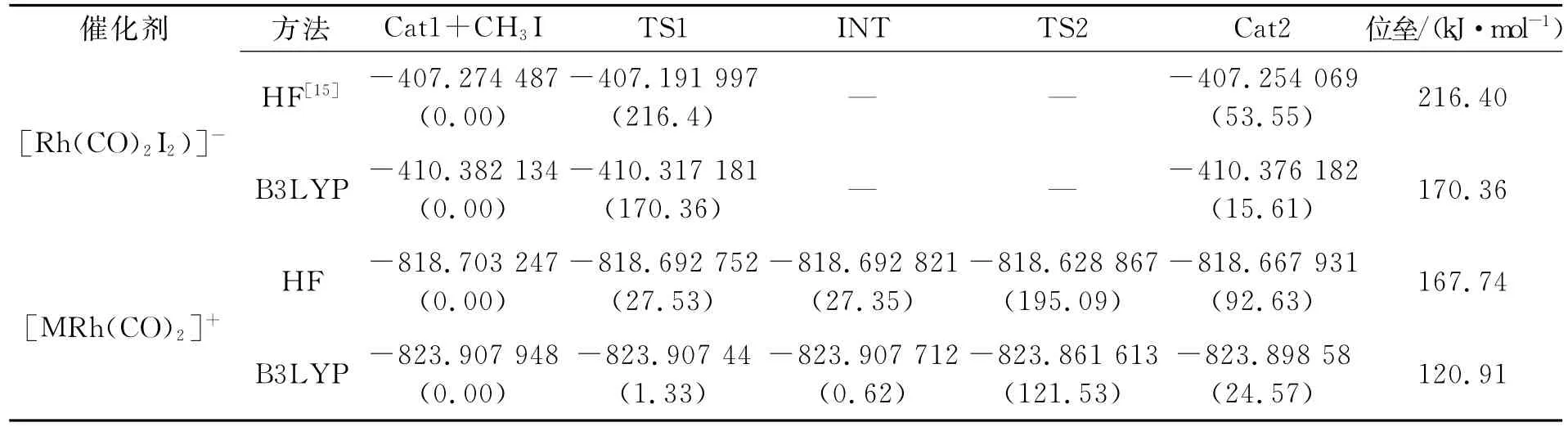
表1 [Rh(CO)2I2)]-和[MRh(CO)2]+催化剂CH3I氧化加成反应中各驻点的能量(包含零点校正能)(单位:Hartree)及相对能量(括号内,单位:kJ·mol-1)
2.3 HOMO、LUMO轨道轮廓、轨道能级分布及催化剂构型分析采用HF方法和B3LYP泛函在赝势基组LANL2DZ下计算并分析了[Rh(CO)2I2)]-和[MRh(CO)2]+的分子轨道能级分布(图3),可以看出,HOMO(最高占据轨道)和LUMO(最低空轨道)的能级差[MRh(CO)2]+为3.27eV,小于[Rh(CO)2I2)]-的3.90eV,可知相比之下[MRh(CO)2]+与CH3I氧化加成时更具有反应活性,这也与前面的结果相一致.我们进一步分析了[MRh(CO)2]+的前线轨道(图4),从前线轨道的轮廓图上可以看出,LUMO轨道具有比较大的离域效应,值得注意的是,除了吡啶环上的离域π键以外,C2—C11之间也形成了很大的离域π键,产生了共轭结构,使得C2—C11具有了双键的性质.这种性质同样从该催化剂的构型上也可以看出来(图1Cat1),C2—C11键长为0.148nm,介于典型的单键键长(0.154nm)和典型的双键键长(0.133nm)之间[17],这实际上是C2—C11与吡啶环之间形成了一个共轭体系,使得π键离域化,C2—C11键具有了双键性质的结果.此时C2—C11键不能够自由旋转,或者说反应时C2—C11键旋转需要较大能量破坏共轭结构.同时,由于邻位的吡啶甲酸与铑配位后,中心原子Rh与周围的基团形成一个四配位平面结构,中心原子Rh与N10、O12、C2、C11形成一个五元环状结构,这是一个难于变形的刚性结构,加之C2—C11键的双键性质,不能旋转,导致决速步骤CH3I与[MRh(CO)2]+的氧化加成反应过程中,五元环无法利用自身的结构调整来协同催化剂的结构挠变,从而加大了反应位阻,导致位垒升高.但是这也同时为该催化剂的改进研究指明了方向,即改变催化剂构型,破坏刚性五元环结构,同时避免C2—C11间离域π键的形成.
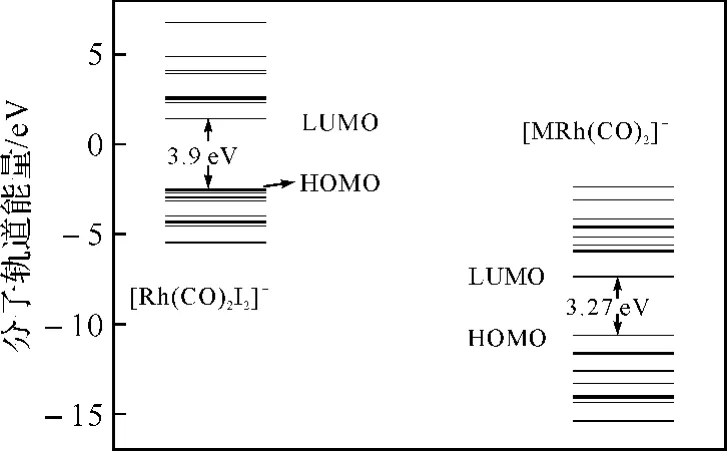
图3 [Rh(CO)2I2)]- 和[MRh(CO)2]+ 的分子轨道能级分布
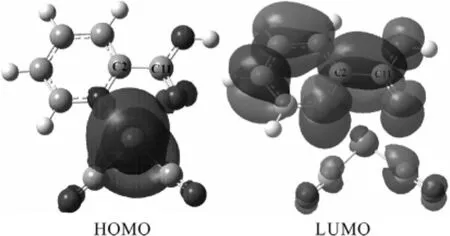
图4 [MRh(CO)2]+的分子轨道轮廓
3 结论
采用密度泛函方法(DFT),在B3LYP/LANL2DZ水平上,参考前期研究工作结论[11],通过决速步骤CH3I氧化加成反应各驻点能量、反应位垒以及催化剂分子轨道能级分布分析可知,[MRh(CO)2]+催化的位垒(120.91kJ/mol)低于[Rh(CO)2I2)]-催化剂(170.36kJ/mol),对于甲醇羰基化制备乙酸的反应[MRh(CO)2]+具有更高的催化活性,这与实验结果一致[8].IRC和反应各驻点构型表明,[MRh(CO)2]+催化循环中CH3I氧化加成步骤分为两步,期间伴随Rh-O12键的开合以及Rh15-N10-O12平面与Rh15-C16-C17平面之间相对位置的调整,是反应中催化剂结构上的挠变.结合IRC和驻点构型,进一步研究[MRh(CO)2]+催化剂的精细结构和分子轨道轮廓图发现,催化剂以Rh为中心的刚性五元环结构和C2—C11间离域π键的形成不利于协同反应中的挠变,是该催化剂在结构上的缺陷,会导致整个催化循环中决速步骤CH3I氧化加成反应位垒升高,由此推断,改变[MRh(CO)2]+催化剂构型,破坏刚性五元环结构,同时避免C2—C11间离域π键的形成,是提高同类型催化剂活性的有效途径.
[1]王玉和,贺德华,徐柏庆.甲醇羰基化制乙酸[J].化学进展,2003,15(3):215-221.
[2]Jiang H,Dao K S,Pan P l,et al.A new glass of rhodium complexes containing free donor atoms and their intramolecular substitution reaction[J].Chin J Chem,2000,18:752-755.
[3]Paulik F E,Roth J F.Catalysis for the low-pressure carbonylation of methanol to acetic acid[J].Chem Commun,1968,24:1578-1581.
[4]Li F B,Huang J,Zou J,et al.Polymer-derived carbon-supported group VIII metals catalysts for vapor phase carbonylation of methanol[J].Appl Catal A,2003,251(2):295-304.
[5]Qian Q L,Li F B,Yuan G Q.Promoting effect of phosphates upon homogeneous methanol carbonylation[J].Catal Commun,2005,6(7):446-448.
[6]Zhang Sh F,Guo C Y,Qian Q L,et al.Synthesis of acetic acid and acetic anhydride from methanol carbonylation with polymer supported rhodium catalyst[J].Catal Commun,2008,9(5):853-858.
[7]Sarmah B J,Borah B J,Deb B,et al.Dicarbonylrhodium(I)complexes of pyridine alcohol ligands and their catalytic carbonylation reaction[J].J Mol Catal A,2008,289(1-2):95-99.
[8]张抒峰,刁开盛,邹瑾,等.吡啶甲酸锂-铑(Ⅰ)配合物催化甲醇羰基化反应[J].化学通报,2001,64(12):781-784.
[9]潘平来,柳忠阳,黄茂开,等.铑配合物催化甲醇羰基化反应的性能和机理[J].催化学报,1995,17(1):45-49.
[10]袁国卿,潘平来,柳忠阳,等.正方平面顺二羰基(三齿N-配体)铑阳离子配合物的分子内取代反应[J].中国科学(B辑),1996,26(2):118-123.
[11]吉文欣,刘翔宇,冀永强,吡啶甲酸铑阳离子催化甲醇羰基化反应机理的理论计算[J].催化学报,2009,30(11):1096-1100.
[12]孙少学,鲁云洲,石从云,等.氟代次甲基与氧气的反应机理研究[J].湖北大学学报:自然科学版,2010,32(2):178-205.
[13]郝茂荣,铑碘催化甲醇羰基化[D].北京:北京化工大学,2003.
[14]郝茂荣,冯文林,冀永强,等.铑碘催化剂催化甲醇羰基化反应的IRC解析[J].中国科学(B辑),2003,33(4):287-295.
[15]雷鸣,冯文林,郝茂荣,等.甲醇羰基化制乙酸反应的理论研究[J].中国科学(B辑),2001,31(5):462-467.
[16]Frisch M J,Trucks G W,Schlegel H B,et al.Gaussian 03(Revision B.04).Pittsburgh(PA):Gaussian Inc,2003.
[17]温建辉.烃分子中碳碳键长的变化[J].大学化学,1992,7(2):52-56.
Theoretical study for geometries,IRC and catalytic performance of methanol carbonylation over pyridinecarbonylic acid rhodium cation
JI Wenxin,LIU Xiangyu,JI Yongqiang
(Key Laboratory of Energy Source and Chemical Engineering,Ningxia University,Yinchuan 750021,China)
Density functional theory(DFT)andabinitioHartree-Fock(HF)had been performed on rate-determining step of methanol carbonylation over pyridine carbonylic acid rhodium cation([MRh(CO)2]+,where M=pyridine carbonylic acid ligand).All structural geometries of reactant,intermediates,transition states and product,respectively,were optimized at in the HF/LANL2DZ and B3LYP/LANL2DZ level.Next,changes of bond lengths of interacting molecules were analyzed along the minimum energy paths(MEP)with the intrinsic reaction coordinate(IRC)theory.Finally,by analyzing molecule obit distributing and geometry,the limitation of[MRh(CO)2]+hed been reached.The results indicated that[MRh (CO)2]+had been better catalytic active than[Rh(CO)2I2)]-.But the formation and characteristic of the rigid 5-membered ring and delocalization πbond between C2and C11was a disadvantage for it.
Hartree-Fock;density functional theory(DFT);Rh;catalysis;geometry
O643
A
1000-2375(2011)04-0505-05
2011-03-21
宁夏大学科学研究基金项目(NDZR10-23)资助
吉文欣(1977-),男,讲师,E-mail:jwx@nxu.edu.cn
(责任编辑 胡小洋)
猜你喜欢
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
茶叶通讯(2019年3期)2019-02-16
浙江化工(2019年5期)2019-01-20
中国园林(2018年7期)2018-08-07
中成药(2017年7期)2017-11-22
北京航空航天大学学报(2017年10期)2017-04-20
党员电教与远程教育(2016年3期)2016-03-19
党的生活·党员电教与远程教育(2016年3期)2016-02-26
源流(2015年8期)2015-09-16
航天返回与遥感(2014年4期)2014-07-31
