
新型含杂环醚类手性配体的合成及其不对称催化性能*
2014-06-23 16:22王丽丽姜艳
合成化学 2014年3期
王丽丽,姜艳
(常州大学石油化工学院,江苏常州 213164)
新型含杂环醚类手性配体的合成及其不对称催化性能*
王丽丽,姜艳
(常州大学石油化工学院,江苏常州 213164)
以L-酒石酸和二苯甲酮为原料,经酯化、缩酮、还原等反应合成了两个新型的含杂环醚类手性配体——(4S,5S)-5-甲基(2-甲氧基吡啶)-2,2-二苯基-1,3-二氧戊环-4-甲醇(7)和(4S,5S)-2,2-二苯基-1,3-二氧戊环-4,5-二甲基(2-甲氧基吡啶)(8),其结构经1H NMR,13C NMR和ESI-MS表征。以环己酮与对硝基苯甲醛的不对称aldol反应为探针反应,考察了7和8的不对称催化活性。实验结果表明,在最佳反应条件[8 0.015 mmol,在甲苯中于10℃反应96 h]下,8的催化效果较佳(ee值97.2%)。
L-酒石酸;手性配体;合成;不对称催化性能
酒石酸是具有C2对称结构的天然手性源,其经典衍生物如DIOP和TADDOL在不对称催化反应中的不对称诱导效果显著[1]。因此,酒石酸及其衍生物在不对称合成领域备受关注并得到广泛应用。Kagan等[2]以天然酒石酸为原料成功合成了手性双膦配体DIOP,拉开了对手性配体的合成研究的帷幕;Seebach等[3]将TADDOL的羟基进行取代,合成了一系列新的酒石酸衍生物并研究其不对称催化性能,产物的ee值达84%。Cisnetti[4]曾报道醚类配体成功与高氯酸钴成功配位。
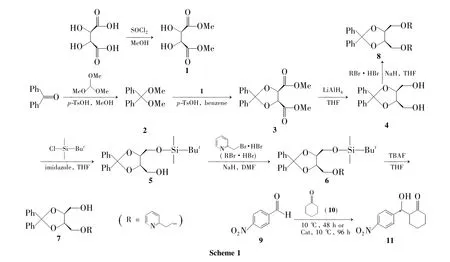
本课题组也合成了类似的配体并进行了改进,保留一个活性羟基,同时与吡啶环上的N原子和醚链骨架上的O原子参与配位。本文在前期研究[5]的基础上,本文以L-酒石酸为原料,经酯化、缩酮、还原等反应合成了两个新型的醚类手性配体——(4S,5S)-5-甲基(2-甲氧基吡啶)-2,2-二苯基-1,3-二氧戊环-4-甲醇(7)和(4S,5S)-2,2-二苯基-1,3-二氧戊环-4,5-二甲基(2-甲氧基吡啶) (8)(Scheme 1),其结构经1H NMR,13C NMR和ESI-MS表征。并以对硝基苯甲醛(9)与环己酮(10)的不对称aldol反应为探针反应(Scheme 1),考察了7和8的不对称催化活性。
1 实验部分
1.1 仪器与试剂
Bruker AVANCE 300 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标);LCMS-2010EV型高效液相色谱-质谱联用仪(LC-MS)。
其余所用试剂均为分析纯。
1.2 合成
(1)酒石酸二甲酯(1)的合成[6]
在干燥三口瓶中加入L-酒石酸30.0 g(199.88 mmol)和甲醇25.6 mL,冰浴冷却下缓慢滴加氯化亚砜80.0 g(672.27 mol),滴毕,撤去冰浴,回流(62℃)反应至终点[TLC检测,展开剂:A=V(乙酸乙酯)∶V(石油醚)=1∶1,碘缸显色,下同]。减压蒸除氯化亚砜,于60℃旋蒸脱溶得淡黄色透明黏稠液体1 33.7 g(低温为白色固体),收率94.8%,m.p.46.3℃~47.2℃。
(2)二甲氧基二苯基甲烷(2)的合成[7]
在干燥三口瓶中加入二苯甲酮4.0 g(21.95 mmol),原甲酸三甲酯5.0 mL(45.59 mmol),对甲苯磺酸(p-TsOH)0.2 g(0.12 mmol)和甲醇12 mL,搅拌使其溶解;回流反应至终点(析出白色固体)(TLC检测,展开剂:A=1∶15)。冷却,抽滤,滤饼用甲醇洗涤后干燥得白色固体2 4.9 g,收率97.7%,m.p.102.7℃~103.5℃。
(3)(4S,5S)-2,2-二苯基-1,3-二氧戊环-4,5-二甲酸二甲酯(3)的合成[8]
在干燥三口瓶中加入2 8.1 g(35.48 mmol),苯200 mL,1 7.6 g(42.7 mmol)和p-TsOH 0.4 g (0.24 mmol),搅拌下回流(80℃)反应至终点(TLC检测,展开剂:A=1∶10)。常压蒸馏除去苯和甲醇共沸物,用三乙胺调至pH 7,于60℃旋蒸脱溶,滴加正己烷2 mL,缓慢加入无水乙醇30 mL(析出白色固体)。过滤,滤饼用无水乙醇重结晶得白色针状固体3 6.6 g,收率54.3%,m.p.80.2℃~80.6℃;1H NMR δ:7.54(m,4H,ArH),7.34~7.29(m,6H,ArH),4.97(s,2H,OCH),3.68(s,6H,CH3);13C NMR δ:168.10,139.16,127.71,127.00,125.64,112.21,76.47,51.63。
(4)(4S,5S)-2,2-二苯基-1,3-二氧戊环-4,5-二甲醇(4)的合成[9]
冰盐浴冷却(-5℃),在干燥三口瓶中加入3 3.0 g(8.76 mmol)和THF 60 mL,搅拌使其溶解;缓慢加入LiAlH41.4 g(36.84 mmol),于0℃反应至终点(TLC检测,展开剂:A=1∶1)。抽滤,滤饼用乙酸乙酯洗涤,合并滤液与洗液,于40℃旋蒸脱溶得白色固体4 2.40 g,收率95.7%,m.p.92.7℃~93.0℃;1H NMR δ:7.50~7.48 (m,4H,ArH),7.36~7.31(m,6H,ArH),4.24(s,2H,OCH),3.81(dd,J=1.2 Hz,1.6 Hz,2H,CH2),3.72(dd,J=1.6 Hz,2.4 Hz,2H,CH2);13C NMR δ:141.64,127.43,127.38,127.29,125.50,125.20,108.76,78.42,61.20; ESI-MS m/z:287.15{[M+H]+}。
(5)(4S,5S)-5-[(叔丁基二甲基)甲氧基]硅烷-2,2-二苯基-1,3-二氧戊环-4-甲醇(5)的合成[10]
于-10℃在干燥三口瓶中依次加入4 0.72 g (2.50 mmol),咪唑0.17 g(2.50 mmol)和无水THF 10 mL,搅拌使其溶解;于-10℃缓慢滴加叔丁基二甲基氯硅烷0.19 g(1.25 mmol)的THF(5 mL)溶液,滴毕(析出白色固体),反应至终点(出现双取代,TLC检测,展开剂:A=1∶5)。抽滤,滤液旋掉大部分溶剂,残余物(约1 mL)用水(10 mL)洗涤,石油醚25 mL萃取,有机相旋蒸脱溶得白色液体5 0.19 g,收率37.7%;1H NMR δ: 7.48(d,J=7.6 Hz,4H,ArH),7.33~7.29(m,6H,ArH),4.14~4.11(m,1H,OCH),4.09~4.06(m,1H,OCH),3.97~3.93(m,1H,CH2),3.77~3.74(m,2H,CH2),3.61~3.57 (m,1H,CH2),0.86(s,9H,CH3in Bu),0.07 (s,6H,CH3);13C NMR δ:143.64,129.37,128.86,128.39,128.20,128.11,118.36,81.32,80.24,60.57,26.32,2.27。
(6)(4S,5S)-5-[(叔丁基二甲基)甲氧基]硅烷-2,2-二苯基-1,3-二氧戊环-4-甲氧基甲基吡啶(6)的合成
在干燥三口瓶中加入5 1.0 g(2.50 mmol)和DMF 15 mL,搅拌下缓慢加入氢化钠0.30 g(12.5 mmol),搅拌使其溶解;缓慢加入溴甲基吡啶溴酸盐(RBr·HBr)0.63 g(2.50 mmol),于室温反应至终点(TLC检测)。加入水15 mL猝灭反应,用乙酸乙酯(3×20 mL)萃取,合并有机相,用无水Na2SO4干燥,浓缩后经硅胶柱层析(洗脱剂:A= 1∶2)纯化得白色黏稠液体6 0.66 g,收率53.6%;1H NMR δ:8.54(s,1H,PyH),7.74(s,1H,PyH),7.53~7.48(m,6H,ArH),7.34~7.31 (m,4H,ArH),7.30~7.28(m,2H,PyH),4.76(s,2H,OCH2),4.31~4.19(m,2H,CH2),4.04(d,J=4.4 Hz,1H,CH),3.89~3.85(m,1H,CH),3.73~3.65(m,2H,CH2),0.85(s,9H,CH3in Bu),0.02(s,6H,CH3);13C NMR δ:156.12,148.79,143.66,137.33,129.41,128.84,128.63,128.31,128.24,121.22,118.44,75.13,69.56,64.21,56.13,30.32,26.22,2.33。
(7)7的合成
在干燥单口烧瓶中加入6 0.5 g(1.02 mmol) 和THF 50 mL,搅拌使其溶解;加入四丁基氟化铵(TBAF)2.0 g(7.66 mmol),于室温反应至终点(TLC检测)。于40℃旋蒸除溶,残余物用水20 mL洗涤,用乙酸乙酯(3×30 mL)萃取,合并有机相,浓缩后经硅胶柱层析(洗脱剂:A=2∶1)纯化得淡黄色固体7 0.24 g,收率62.5%,m.p.111.0℃~111.2℃,[α]20D+37.76°(c 0.5,CHCl3,下同);1H NMR δ:8.52(d,J=4.5 Hz,1H,PyH),7.69~7.63(t,J=4.5 Hz,1H,PyH),7.51~7.49 (m,2H,PyH),7.48~7.46(m,2H,ArH),7.34~7.27(m,7H,ArH),7.20~7.16(t,J=6.1 Hz,1H,ArH),4.66(s,2H,OCH2),4.34~4.28 (m,1H,OCH),4.17~4.11(m,1H,OCH),3.85~3.77(m,3H,CH2),3.65~3.59(m,1H,CH2);13C NMR δ:157.69,149.03,142.73,136.81,128.24,128.14,128.09,126.19,126.05,122.55,121.39,109.83,80.95,74.19,71.04,62.50;ESI-MS m/z:378.15{[M+H]+},400.15{[M+Na]+}。
(8)8的合成
在干燥单口烧瓶中依次加入4 0.1 g(0.35 mmol),NaH 91 mg(0.25 mmol)和THF 50 mL,搅拌使其溶解;缓慢加入RBr·HBr 0.19 g(0.75 mmol),于室温反应至终点(TLC检测)。抽滤,滤饼用乙酸乙酯洗涤,合并滤液和洗液,于40℃旋蒸除溶后经硅胶柱层析(洗脱剂:A=3∶1)纯化得白色透明液体8 0.15 g,收率91.5%,[α]20D+12.69°;1H NMR δ:8.52(d,J=4.3 Hz,2H,PyH),7.67~7.61(d,J=4.3 Hz,2H,PyH),7.52(dd,J=4.3 Hz,1.6 Hz,4H,PyH),7.37~7.23(m,6H,ArH),7.19~7.15(m,4H,ArH),4.69(s,4H,CH2),4.29~4.27(t,J=3.0 Hz,2H,OCH),3.80~3.69(m,4H,CH2);13C NMR δ: 157.18,147.95,141.84,135.63,127.04,126.99,125.27,121.33,120.23,109.18,77.46,76.42,76.20,76.00,75.58,73.23,70.18;ESI-MS m/z: 469.15{[M+H]+},491.15{[M+Na]+}。
(9)2-[羟基(4-硝基苯)甲基]环己酮(11)的合成
于10℃在反应瓶中依次加入9 0.755 g(5.0 mmol),10 5 mL(0.05 mol),搅拌下于10℃反应48 h(TLC检测)。旋蒸(30℃)脱溶后经硅胶柱层析(A=1∶5)纯化得淡黄色固体1.09 g,收率87.5%;1H NMR δ:8.21(d,J=8.4 Hz,2H,ArH),7.52(d,J=8.8 Hz,2H,ArH),4.90(d,J=8.4 Hz,1H,CH),4.06(s,1H,OH),2.63~2.56(m,1H,CyH),2.52~2.48(m,1H,CyH),2.41~2.32(m,2H,CyH),2.04~1.85(m,1H,CyH),1.43~1.26(m,2H,CyH)。
以7或8(0.015 mmol)为催化剂,反应时间96 h,其余反应条件同上,制得11,收率分别为72.0%和65.0%。
2 结果与讨论
2.1 合成
(1)1的合成
酯的制备方法较多,可通过羧酸和醇的直接酯化、羧酸盐和活泼卤代烷的取代反应、羧酸的醇解、羧酸与重氮甲烷反应等方法合成。羧酸与醇直接酯化是实验室最常用的方法。
考虑到反应时间、催化剂的选择及副产物单酯,本文采用L-酒石酸与二氯亚砜在低温条件下反应生成酰氯后再与甲醇反应制备1。甲醇既作溶剂又作反应试剂。L-酒石酸有两个活性羟基,为了避免与二氯亚砜及中间体酰氯反应,在低温条件下滴加二氯亚砜,滴加完毕后立即升温至60℃与甲醇反应制得双酯1,后处理十分简便,产率也高(94.8%)。
(2)2的合成
3 是二苯甲酮与1缩酮产物,理论上可由二苯甲酮和1直接反应制得。但在实验过程中,我们尝试分别以苯、甲苯为带水剂,对甲苯磺酸为催化剂,加热回流反应24 h后仍无产物生成。原因是二苯甲酮的空间位阻比较大,反应需要更高的能量才能发生。而先用二苯甲酮与原甲酸三甲酯反应制得2,再与1反应即可快速反应生成产物2。二苯甲酮和原甲酸三甲酯在酸催化作用下反应,由于反应过程中不会产生水,产率较高且后处理简单。但2在空气中不稳定,很容易分解,不能长时间保存。
(3)3的合成
2 和1 反应时,TLC跟踪检测无2,但二苯甲酮点仍然很浓,尝试补加原甲酸三甲酯希望能促进反应平衡的移动,继续加热回流3 h后发现二苯甲酮点没有变淡,即停止反应,说明反应中补加的原甲酸三甲酯不再参与反应。可能是因为点板过程中2遇空气分解。该步反应产物用无水乙醇重结晶,后处理简便。
(4)4的合成
合成4时,以氢化铝锂为还原剂,在低温条件下加入氢化铝锂,防止其遇体系中残留的微量水剧烈反应放出大量热而无法及时散去。于0℃下反应至原料点消失,反应条件温和且速度快,反应彻底,后处理十分简便。
(5)5的合成
在5的合成中,叔丁基二甲基氯硅烷的THF溶液必须冷却至-10℃,否则温度过高很容易生成双取代产物,滴加过程中出现的白色固体,经核磁验证为反应物二醇。反应过程中需要用TLC连续跟踪,一旦有双取代产物生成即立即停止反应。用水洗涤除去原料二醇,产物用石油醚萃取,后处理简单,原料二醇可回收再利用。
(6)6的合成
在6的合成中,由于RBr·HBr相对比较贵,5应过量,反应跟踪至溴甲基吡啶的点消失时停止反应。
(7)7的合成
TBAF是脱硅醚保护的一种良好试剂,一般在中性至弱酸性条件下脱保护。本文采用无水THF为溶剂,否则TBAF遇水会生成强碱,强碱会影响反应进程。
(8)8的合成
用无水THF为溶剂,NaH先与醇生成强碱醇钠作为缚酸剂。当RBr·HBr加完后立刻TLC跟踪检测,观察发现同时有两个新点生成,随着反应进行,单取代点慢慢变淡,最后几乎没有。而双取代点逐渐变浓,产率较高。理论上单取代产物的极性应该比双取代产物大,因为其含有一个极性较大的羟基,但是双取代产物的极性要大于单取代产物,原因是单取代产物分子结构中的吡啶环上的N原子与羟基发生某种氢键作用导致整个分子极性减小。
由于反应在室温条件下进行,且将RBr·HBr一次加完,导致双取代产物很快生成。4是伯醇,选择性比较差,如果要使单取代产物为主产物,减少双取代产物的生成,尝试低温反应,并将RBr· HBr以溶液形式慢慢滴加到反应液中,使反应液浓度降低,尽量降低分子碰撞几率以提高单取代产物比例,但实验证明双取代产物仍为主产物。
2.2 7和8的不对称催化性能
7和8的不对称催化性能见表1。由表1可见,在无催化剂条件下,所得产物11为消旋体,产率87.5%;以7或8为催化剂时所得11具有一定ee值,表明7和8对该不对称aldol反应具有较好的催化效果。8含有一个活性羟基、具有不对称结构,其催化效果较佳(ee值97.2%)。
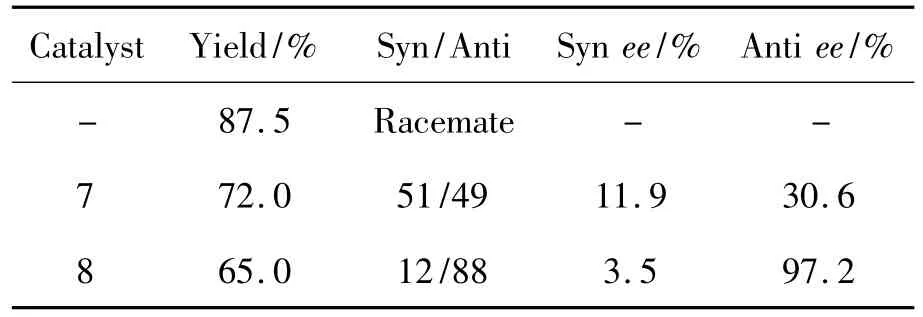
表1 环己酮和对硝基苯甲醛的不对称aldol反应Table 1Asymmetric aldol reaction of cyclohexanone with p-nitrobenzaldehyde
3 结论
以L-酒石酸和二苯甲酮为原料,合成了两个新型的带吡啶环的醚类手性配体——(4S,5S)-5-甲基(2-甲氧基吡啶)-2,2-二苯基-1,3-二氧戊环-4-甲醇(7)和(4S,5S)-2,2-二苯基-1,3-二氧戊环-4,5-二甲基(2-甲氧基吡啶)(8)。手性中心与配位原子之间有亚甲基,增加了配体的柔性,可根据与金属的配位构型变化以保证配合物的稳定性。7和8对环己酮与对硝基苯甲醛的不对称aldol反应具有较好的催化效果。8的催化效果较佳(ee值97.2%)。
[1]韩玉峰,沈宗旋,蒋虹,等.手性催化剂(4R)-苄氧基-(S)-脯氨酸的合成及其对不对称羟醛反应的催化活性[J].应用化学,2004,21(6):641-644.
[2]Kagan H B,Dang-Tuan-phat.Asymmetric catalytic reduction with transition metal complexes.Ⅰ.Cataytic system of rhodium(Ⅰ)with(-)-2,3-O-isopropylidene-2,3-dihydroxy-1,4-bis(diphenylpho-sphino)butane,a new chiral diphosphine[J].Journal of the A-merican Chemical Society,1972,94(18):6429-6433.
[3]Dieter Seebach,Albert K Beck,Alexander Heckel. TADDOLs,their derivatives,and TADDOL analogues: Versatile chiral auxiliaries[J].Angew Chem Int Ed,2001,40:92-138.
[4]Federico Cisnetti,Régis Guillot,Michel Thérisod,et al.Diaqua[methyl 4-O-benzyl-2,3,6-tri-O-(2-picolyl)-α-D-mannopyranoside〛cobalt(Ⅱ)bis(perchlorate)monohydrate:Partial coordination of a ligand derived from D-mannose[J].Acta Crystallographica Section C,2007,C63:m201-m203.
[5]Jiang Y,Bian J,Sun X.[(4S,5S)-2,2-Dimethyl-1,3-dioxolane-4,5-diyl]bis[N-(thiophen-2-ylme-thylidene)-methanamine][J].Acta Cryst,2012,E68: o430-o430.
[6]Tsujimoto T Ito Y.Concise syntheses of immunostimulating glycolipids,α-galactosyl c-eramides[J].Tetrahedron Letters,2007,48(31):5513-5516.
[7]Belen Altava,M Isabel Burguete,Eduardo Garcia-Verdugo,et al.On the origin of changes in topicity observed in Diels-Alder reactions catalyzed by Ti-TADDOLates[J].Tetrahedron Asymmetryt,2000,4885-4893.
[8]Altava B,Burguete M I,Eduardo G,et al.On the origin of changes in topicity observed in Diels-Alder reactions catalyzed by Ti-TADDO Lates[J].Tetrahedron:Asymmetry,2000,11:4885-4893.
[9]MaGee D I,Silk P J,Wu J,et al.Synthesis of chiral alkenyl epoxides:The sex pheromone of the elm spanworm ennomos subsignarius(Hubner)(Lepidoptera: Geometridae)[J].Tetrahedron,2011,67(29):5329-5338.
[10]Corey E J,Venkateswarlu A.Protection of hydroxyl groups as tert-butyldimethylsilyl derivatives[J].J Am Chem Soc,1972,94:6190-6191.
Synthesis and Asymmetric Catalytic Performance of Novel Heterocyclic Ethers Chiral Ligands
WANG Li-li,JIANG Yan
(School of Petrochemical Engineering,Changzhou University,Changzhou 213164,China)
Two novel heterocyclic ether chiral ligands,{(4S,5S)-2,2-diphenyl-5-[(pyridin-2-ylmethoxy)methyl]-1,3-dioxolane-4-yl}methanol(7)and 2,2'-【{[(4S,5S)-2,2-diphenyl-1,3-dioxolane-4,5-diyl]bis(methylene)}bis(oxy)bis(methylene)】dipyridine(8),were synthesized by esterification,ketal and reducation reactions using L-tartaric acid and benzophenone as the raw materials. The structures were characterized by1H NMR,13C NMR and ESI-MS.The asymmetric catalytic performance of 7 and 8 were investigated by aldol reaction of cyclohexanone with p-nitrobenzaldehyde. The results showed that 7 and 8 exhibited certain catalytic performance,and under the optimum reaction conditions[8 0.015 mmol,at 10℃for 96 h,toluene as the solvent],the catalytic performance of 8 is better with ee value of 97.2%.
L-tartaric acid;chiral ligands;catalytic;synthesis;asymmetric catalytic performance
O626;O621.3
A
1005-1511(2014)03-0308-05
2013-03-13;
2014-03-05
国家自然科学基金资助项目(20872051)
王丽丽(1988-),女,汉族,江苏常州人,硕士研究生,主要从事有机合成和不对称羟醛缩合反应的研究。E-mail:erliwanglili@163.com
姜艳,副教授,E-mail:jy@cczu.edu.cn
猜你喜欢
电镀与精饰(2022年3期)2022-03-14
长江大学学报(自科版)(2021年3期)2021-06-01
理化检验-化学分册(2020年5期)2020-06-15
电脑报(2020年10期)2020-04-28
山东化工(2019年7期)2019-04-27
中国资源综合利用(2017年1期)2018-01-22
中国塑料(2017年2期)2017-05-17
上海蔬菜(2015年2期)2015-12-26
苏州科技大学学报(工程技术版)(2015年3期)2015-02-28
安徽医药(2014年4期)2014-03-20
