
7β-苯乙酰胺-3-头孢美法仑-4-头孢烷酸的合成*
2014-06-23 16:22周晓靓王浩李卫红施培基周则卫
合成化学 2014年3期
周晓靓,王浩,李卫红,施培基,周则卫
(中国医学科学院放射医学研究所药物室,天津 300192)
·制药技术·
7β-苯乙酰胺-3-头孢美法仑-4-头孢烷酸的合成*
周晓靓,王浩,李卫红,施培基,周则卫
(中国医学科学院放射医学研究所药物室,天津 300192)
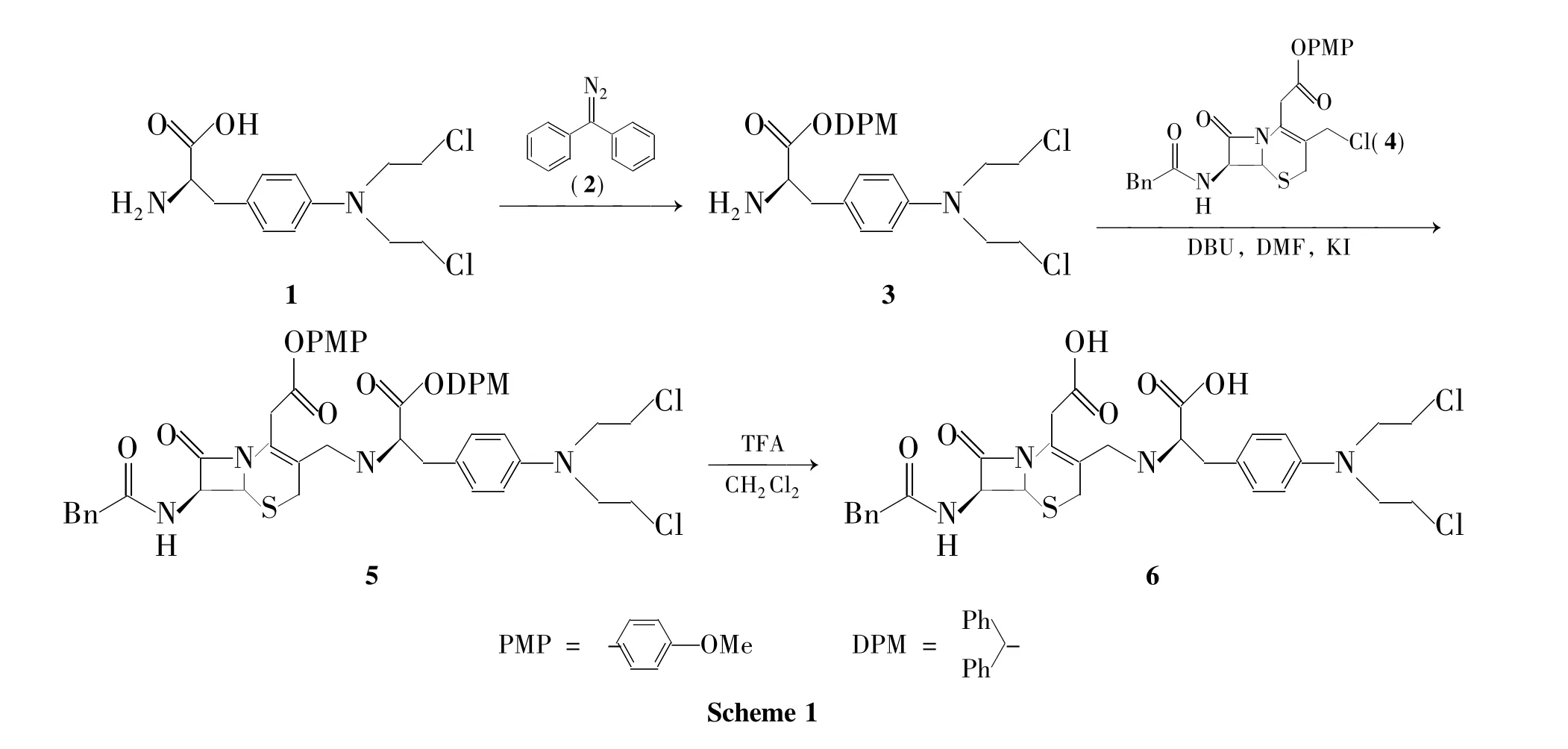
美法仑与二苯基重氮甲烷经回流反应制得美法仑二苯甲酯(3);首次采用7-苯乙酰氨基-3-氯甲基-4-头孢烷酸对甲氧基苄酯与3在DBU作用下经“一锅法”反应后水解合成了前体药物头孢美法仑(7β-苯乙酰胺-3-头孢美法仑-4-头孢烷酸),其结构经1H NMR和ESI-MS确证。
前药;头孢菌素;美法仑;药物;合成
化疗是当前治疗恶性肿瘤的重要手段之一。如何增加化疗药物在肿瘤的局部浓度同时降低全身暴露浓度是当前肿瘤靶向治疗的热点。抗体导向酶解前药治疗(ADEPT)可在增加靶细胞药物浓度的同时降低正常组织的药物浓度,减少化疗引起的毒副作用并提高疗效。在ADEPT研究系统中,根据β-内酰胺酶的生化特点和头孢菌素的化学结构特点,选择β-内酰胺酶为活化酶,头孢菌素拼合细胞毒化合物作为底物,在ADEPT研究中较为常见[1-2]。头孢美法仑(6)是将头孢菌素与氮芥类非特异性抗肿瘤药物拼合而成的前体药物,在β-内酰胺酶的特异性作用下可以转化为原型药物美法仑,从而发挥抗肿瘤作用。
文献[3]方法中,6的合成以7-ACA为原料制得重要活性中间体7-羟甲基头孢后,再与美法仑(1)进行偶联而得。该方法合成步骤多,涉及多个活性基团的保护和去保护。
本文以1与二苯基重氮甲烷(2)经回流反应制得美法仑二苯甲酯(3)后;首次采用7-苯乙酰氨基-3-氯甲基-4-头孢烷酸对甲氧基苄酯(GCLE,4)为原料,与3在1,8-二氮杂二环[5.4.0]十一碳-7-烯(DBU)作用下经“一锅法”反应后水解合成了用于肿瘤靶向治疗的前药6(Scheme 1),其结构经1H NMR和ESI-MS确证。
1 实验部分
1.1 仪器与试剂
Varian Mercury Vx-300型核磁共振仪(CDCl3为溶剂,TMS为内标);WATERS ALLIANCE型高效液相色谱仪[ODS柱(4.5 mm×250 mm),流动相:甲醇/乙酸胺缓冲液/乙酸=80/20/1,流速: 1.0 mL·min-1,检测波长:254 nm]。
2 按文献[4]方法合成;1,苏州立德化学有限公司;4,天津津康药业有限公司;DBU,北京偶合科技有限公司;其余所用试剂均为分析纯。
1.2 合成
(1)3的合成
在反应瓶中依次加入丙酮30 mL和1 1.2 g (3.9 mmol),搅拌使其呈混悬液;缓慢加入新制的2(12 mmol)的正己烷(12 mL)溶液,加毕,回流反应2 h[TLC监测,展开剂:A=V(乙酸乙酯)∶V(环己烷)=4∶1]。过滤,滤液减压浓缩后经加压硅胶柱层析(梯度洗脱剂:A=1∶1~1∶0)纯化得黄色油状物3 1.4 g,收率76%,Rf=0.2(展开剂:A=1∶1);1H NMR δ:7.36~7.22(m,10H),6.94~6.90(m,2H),6.49~6.47(d,J=8.4 Hz,2H),3.84~3.79(m,1H),3.70~3.55(m,8H), 3.06~3.00(dd,J=5.7 Hz,5.4 Hz,1H),2.90~2.83(dd,J=6.9 Hz,7.2 Hz,1H),1.94(br,2H)。
(2)7β-苯乙酰胺-3-头孢美法仑-4-头孢烷酸对甲氧基苄酯(5)的合成
在反应瓶中依次加入DMF 15 mL和4 1.0 g (2 mmol),搅拌使其呈混悬液;加入3 1.3 g(2 mmol),于-5℃加入碘化钾0.8 g(4.8 mmol),缓慢滴加DBU 600 μL,滴毕,避光反应5 h(TCL监控,反应液颜色逐渐加深)。用乙酸乙酯(3×30 mL)萃取,合并萃取液,依次用10%硫代硫酸钠溶液、10%柠檬酸水溶液和饱和食盐水洗涤,无水硫酸镁干燥,减压浓缩后经加压硅胶柱层析(梯度洗脱剂:A=1∶1~1∶0)纯化得淡黄色固体5 750 mg,收率40%,Rf=0.35(展开剂:A=1∶1);1H NMR δ:7.37~7.26(m,18H),7.01~6.98 (d,J=5.7 Hz,2H),6.80(s,2H),6.52~6.49 (d,J=9.0 Hz,2H),6.05~6.02(d,J=9.0 Hz,1H),5.85~5.80(m,1H),5.35~5.34 (dd,J=4.2 Hz,5.7 Hz,1H),4.89~4.88(d,J=5.1 Hz,1H),4.12~3.55(m,16H),3.21~3.15(dd,J=8.4 Hz,9.6 Hz,2H),1.70(s,2H);ESI-MS m/z:920.291 1[M+]。
(3)6的合成
在反应瓶中依次加入二氯甲烷5 mL和5 400 mg(0.43 mmol),搅拌使其溶解;加入苯甲醚1 mL,冰浴冷却下缓慢滴加三氟乙酸(TFA)1 mL(5 min),滴毕,于室温反应30 min。倾入50 mL冷的异丙醚(析出淡黄色固体),离心(2 500 rpm,10 min)分离得5的三氟乙酸盐,经硅胶柱层析(洗脱剂:乙醇)纯化得类白色固体6 120 mg,含量93.53%(T=3.341 min),Rf=0.75[展开剂:V(乙醇)∶V(乙酸)=10∶1];UV:259.7 nm,301.3 nm;1H NMR δ:8.51(s,1H),8.21(d,J=8.3 Hz,1H),7.31~7.30(m,5H),6.97(s,2H),6.59~6.56(d,J=8.7 Hz,2H),5.61~5.59(dd,J=4.8 Hz,1H),5.12~5.07(d,J=13.8 Hz,1H),4.82~4.80(d,J=4.8 Hz,1H),4.58~4.54(d,J=13.3 Hz,1H),4.05~4.01(m,1H),3.76~3.21(m,12H),3.00(dd,1H),2.70(dd,1H);ESI-MS m/z:631.27{[M-3H]3-}。
2 结果与讨论
2.1 合成
文献报道6的合成以7-ACA为原料,需要合成主要中间体3-羟甲基头孢烷酸,然后与1,2,2,2-四氯乙基氯甲酸(Ⅰ)反应制得活性中间体3-四氯甲基氯甲酸酯,最后与1的氨基反应合成偶联化合物。该路线步骤长,Ⅰ为进口试剂,获得困难。本文以4为原料,利用DBU“一锅法”反应制得偶联化合物,大大缩短了实验路线。
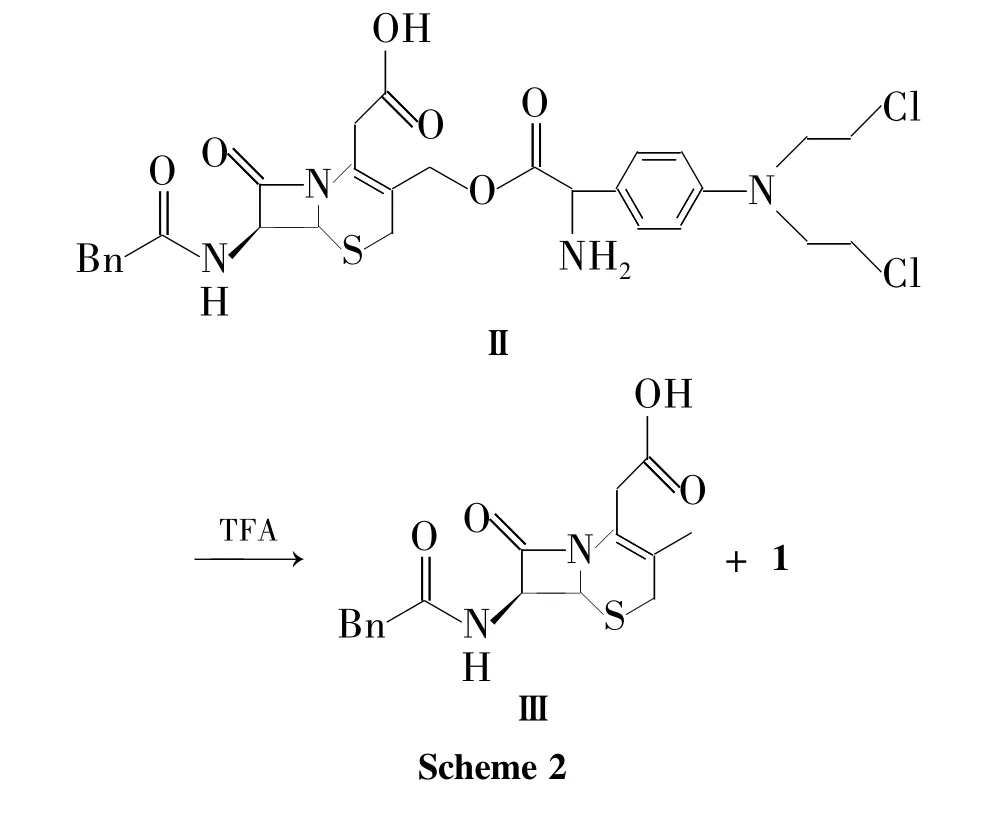
在头孢美法仑的偶联反应(3→5)中,对1的羧酸成酯进行保护,如果使用未保护的1进行偶联反应,所得优势产物为头孢菌素3-位氯甲基与1的羧酸反应成酯产物(Ⅱ),在TFA水解过程中酯键会断裂生成3-甲基头孢菌素(Ⅲ)和1 (Scheme 2)。
头孢菌素衍生物合成难度主要表现为对酸碱不稳定,反应产物难于重结晶,纯化困难等。本路线中的4在碱性条件下易发生2-位和3-位的双键易位[5],因此偶联反应中的缚酸剂的选择很关键。常用的K2CO3和三乙胺等碱均可能带来双键易位的副产物,难以通过柱层析分离。而DBU则能够避免双键易位,制得较为单一的产物。
本文首次通过一锅法以4为原料合成了头孢美法仑前药6,以DBU为缩合剂避免了副产物生成,简化了合成路线。
该路线也可能用于其他头孢菌素衍生物的合成改造,为肿瘤靶向治疗ADEPT的研究提供了化学基础。
[1]David E Kerr,Zhengong Li,Nathan O Siemers,et al.Development and activities of a new melphalan prodrug designed for tumor-selective activation[J].Bioconj Chem,1998,2:255-259.
[2]Ralph F Alderson,Brian E Toki,Roberge M,et al.Characterization of a CC49-based single-chain fragment-β-lactamase fusion protein for antibody-directed enzyme prodrug therapy(ADEPT)[J].Bioconj Chem,2006,17:410-418.
[3]Brian E Toki,Peter D Senter.Melphalan Prodrug [P].US 20 050 214 310,2005.
[4]吕茜茜,周晓靓,王荣先.二苯基重氮甲烷的合成[J].化学试剂,2008,30(2):147-147.
[5]Ryan M Phelan,Marc Ostermeier,Craig A Townsend. Design and synthesis of a β-lactamase activated 5-fuorouracil prodrug[J].Bioorg Med Chem Lett,2009,19:1261-1263.
Synthesis of 7β-Phenethyl Carboxamido-3-melphalan-4-cephalosporin
ZHOU Xiao-liang,WANG Hao,LI Wei-hong,SHI Pei-ji,ZHOU Ze-wei
(Pharmceutical department,Institute of Radiation Medicine,Chinese Academy of Medical Sciences,Tianjin 300192,China)
Melphalan diphenylmethyl ester(3)was synthesized by reflux of Melphalan with diphenyldiazomethane.A cephalosporin-mustard prodrug,Cephalosporin melphalan(7β-Phenethyl Carboxamido-3-melphalan-4-cephalosporin),was synthesized by“one-pot”reaction of 7-phenylacetamide-3-chloromethylcephalosporanic acid p-methoxybenzyl ester with 3 and then hydrolysis for the first time. The structure was confirmed by1H NMR and ESI-MS.
prodrug;cephalosporin;Melphalan;synthesis
O621.3;R914.5
A
1005-1511(2014)03-0416-03
2013-12-25;
2014-04-17
国家自然科学基金资助项目(81301983);研究所发展基金资助项目(SF1305)
周晓靓(1979-),女,回族,宁夏石嘴山人,博士研究生,助理研究员,主要从事头孢菌素类靶向前药及辐射防护药的合成研究。Tel.022-85683041,E-mail:zhouxiaoliang@irm-cams.ac.cn
王浩,E-mail:wanghao@irm-cams.ac.cn
猜你喜欢
吉林农业(2019年6期)2019-06-11
心肺血管病杂志(2019年1期)2019-04-22
教育教学论坛(2018年38期)2018-09-25
中成药(2017年10期)2017-11-16
中成药(2017年4期)2017-05-17
河南农业科学(2017年4期)2017-04-12
当代化工研究(2016年9期)2016-03-20
中国塑料(2015年6期)2015-11-13
中国塑料(2015年8期)2015-10-14
中国医药科学(2015年5期)2015-08-01
