
隐马尔科夫模型基于残基对蛋白质序列的分析
2014-07-10 10:42汪一亭
池州学院学报 2014年3期
汪一亭
(池州学院 数学与计算机科学系,安徽 池州247000)
隐马尔科夫模型基于残基对蛋白质序列的分析
汪一亭
(池州学院 数学与计算机科学系,安徽 池州247000)
区分、识别出同源蛋白质序列并揭示不同类型的残基的研究在生物信息领域具有重要的意义。文章将蛋白质的氨基酸与残基的序列用隐马尔科夫模型(HMM)来表示,介绍了一种基于蛋白质残基来建立隐马尔科夫模型的思路。接着采用HMM的评估算法对蛋白质同源性进行分类,又由于是将残基类型作为模型的状态来考虑,利用HMM的结论可以解码出最优的残基序列,从而进一步预测出残基的类型。结果表明分类算法取得了较好的效果,且在预测结果上与其他方法相比也具有一定的优势。
隐马尔可夫模型;蛋白质;界面残基;表面残基
1 引言
近年来,随着蛋白质测序工作的快速发展,人们已经获取了大量蛋白质序列数据。但是,由于同源蛋白质的原因,往往会出现对某类同源的序列进行测序,并将其存入数据库的情况,结果可能会导致对某一簇蛋白质序列的功能分析的夸大,从而对研究产生误导[1]。因此,比较蛋白质序列并区分、识别出同源序列的算法的研究已成为生物信息领域的重要内容。目前,已有不少相关学者对此类问题进行了研究。有聚类算法[2]、基于图论的算法[3]。而隐马尔可夫模型(Hidden Markov model,HMM)用于蛋白质研究是生物信息学研究的新领域,文献 [4]将HMM用于蛋白质同源性的研究,但是该算法建立的HMM模型所基于的特征的关注度和准确性远远没有蛋白质残基的分布特征高。
蛋白质间的相互作用是蛋白质组学研究的另一个核心问题。其中蛋白质界面残基的预测所研究的是确定在蛋白质-蛋白质相互作用中,某一条链上的哪些残基参与了作用[6]。因此捕捉和揭示不同种类的蛋白质残基对深入了解蛋白质间相互作用的机制具有重要的生物学意义。由于生物学实验技术探测残基费时费力且不能大规模应用,近几年已有很多计算方法被提出,主要有SVM方法[7]、贝叶斯方法[6]、神经网络方法[8]等,但相较于传统的分类方法中,利用隐马尔可夫模型预测残基的案例较少。
2 基本理论
2.1 HMM简介
隐马尔可夫模型作为一种统计分析模型,被用于生物信息学研究领域的基础是计算机技术、统计学和分子生物学。它由相互关联的两个随机过程共同描述信号的统计特性,HMM可以用五个元素来描述分别为隐含状态、可观测状态、初始状态概率、隐含状态转移概率矩阵、观测状态概率矩阵。HMM解决的关键实际问题有三类分别是:1)评估问题,采用前向算法;2)解码问题,采用Viterbi算法;3)学习问题,采用Baum-Welch算法。
2.2 蛋白质残基的定义与分类
蛋白质残基是氨基酸序列脱水形成肽链后的部分,共分为界面残基(interface residue)、表面残基(surface residue)、非表面残基(non-surface residue)三类[5]。对于此三类残基的定义还没有完全统一,文献[6]、[10]都有各自的区分残基的标准,但都是基于某一实验手段称为溶剂可及表面积(solvent accessible surface area,SASA)的基础上量化指标的不同而已。其中,界面残基属于表面残基,但比非界面的表面残基具有更高的SASA[9],为方便描述,下文中的表面残基特指非界面的表面残基。三者分布关系见图1。
不仅如此,SASA不仅能区分残基,而且通过SASA还可以了解氨基酸的疏水性,而疏水性对于蛋白质的二级结构预测非常关键。因此对残基的研究无论对于蛋白质间相互作用还是蛋白质空间结构的预测都具有非常关键的意义。我们采用文献[6]的定义:即若残基的SASA对残基的最大面积的比值超过25%,就定义它为表面残基。而在复合物形成过程中SASA的减少量超过1的残基就定义为界面残基。
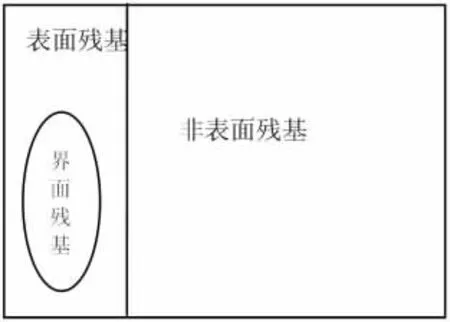
图1 蛋白质残基分布关系
3 HMM模型的建立与算法
3.1 HMM各元素的确立与参数学习
已有学者将HMM用来表示蛋白质序列而进行的研究[4],但该模型的状态是基于进化过程中原始蛋白质序列经历突变,遗失,或引入外源序列,而此类特征的关注度和准确性远远没有蛋白质残基的分布特征高。
在本文中,将蛋白质的氨基酸与残基的序列用HMM模型来表示,其中界面、表面、非表面残基是由难以直接观测到的隐状态表示,构成蛋白质的20种氨基酸由可观测值表示。图2为相应的隐马尔可夫模型。该模型的具体流程是从一个起始隐状态开始,以某种概率进入界面、表面、非表面残基状态之间的某一个,其中每个隐状态会观察到一种氨基酸。当模型从起始状态到结束状态时,产生的不同氨基酸会构成一个氨基酸序列。图中箭头表示的状态间的转换概率是有区别的,此外不同状态所能观测到的氨基酸种类的概率也是有区别的。状态的初始分布、每个状态产生的氨基酸种类的概率、各状态间的转移概率都由模型的参数决定。经过训练,可以调整该模型的参数,训练好的模型能够以最大的可能性产生参与训练的观察序列,由此可以代表有共同特征的蛋白质序列,从而描述不同族的蛋白质。我们将采用典型的HMM训练算法Baumwelch算法来进行建模。
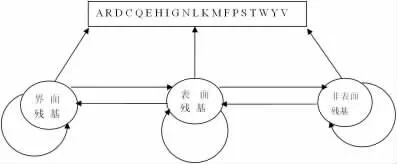
图2 基于蛋白质残基的隐马尔可夫模型
3.2 同源蛋白质序列的区分算法
之后将Baum-welch算法训练出来的参数模型进行蛋白质同源性的研究。其原理为HMM的评估问题,分析由该模型产生不同序列的概率,对于与模型相符合的序列,则能以较大的概率产生该序列,若不与该模型符合的序列,则产生该序列的概率会相对较小,由此可以区分出同源和非同源蛋白质序列,此外,只要对概率划分合适的域值,就能够从许多蛋白质序列中识别出该族的蛋白质序列。我们将采用HMM评估问题的典型算法前向算法来区分蛋白质序列。
3.3 蛋白质残基的预测算法
在上述区分的同源蛋白质的基础上可以进一步根据该模型预测界面、表面、非表面残基,该问题则属于HMM的解码问题。
4 实验结果分析
实验采用的蛋白质复合物为Homo-complex I data set与Hetero-complex I data set[5],这些数据集来源于PDB数据库经过筛选后得到的结果,各自含有621和504条蛋白质复合物链。我们首先从Hetero-complex I data set中选择前50条作为训练序列,经过Baum-welch算法的学习得到隐马尔科夫模型。训练好的该模型参数见表1、2、3。其中,状态0,1,2分别表示界面、表面、非表面残基。

表1 初始状态概率
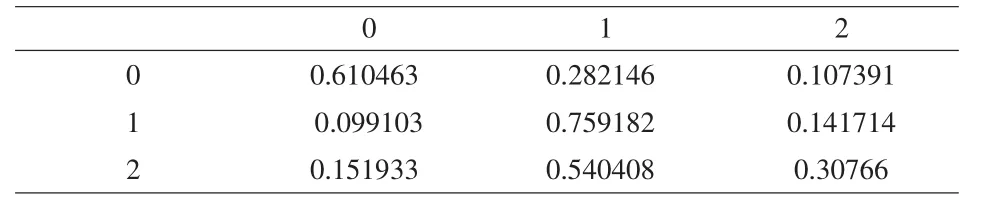
表2 状态转移概率矩阵

表3 氨基酸种类概率矩阵
根据上述参数模型,将Hetero-complex I data set余下的454条蛋白质序列随机选取150条采用前向算法进行分析评估,为了便于统计大量数据结果,程序在实现前向算法的基础上添加计算平均值和方差的功能。再将Homo-complex I data set的蛋白质序列同样方法用该模型进行分析,两次实验各自得到的统计结果见表4。
由表4我们可以清楚看出,模型产生Homocomplex I数据集的蛋白质序列的概率明显小于Hetero-complex I数据集的蛋白质概率。由于这两个数据集分别是描述同类聚合物和异类聚合物的蛋白质序列,由此说明该隐马尔科夫模型能够很好的区分出同源和非同源蛋白质序列。
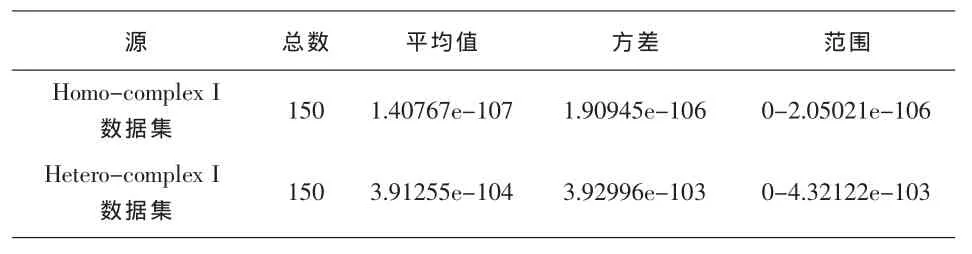
表4HMM评估结果
接下来将与该模型相匹配的余下的蛋白质序列用viterbi算法进行解码,预测出界面、表面、非表面残基的结果见表5,我们选取其中的PDB代码为1xqs蛋白质的D链为例,列举出其部分详细的残基预测结果。其中的T表示该残基为界面残基,S表示该残基为表面残基,N表示非表面残基。残基的序列号是PDB文件中的序列编号,所有残基采用单字母表示。
从表5可以看出,与文献[4]、[6]相比,文献[4]只能识别出同源和非同源蛋白质序列,不能预测残基的种类,而采用我们的方法既能识别出蛋白质序列(如表4所示)又能预测出残基种类;文献[6]采用贝叶斯方法所得到的仅能预测界面残基,而表面残基和非表面残基的情况则没有考虑 (见表6:在1fc2_C链上贝叶斯方法的预测结果),而本文利用的HMM的viterbi算法可以预测出最可能的三类蛋白质残基序列。在预测算法的思想上,传统的分类方法仅分散的研究各残基而忽略相邻残基间的相互关联,本文的马尔科夫模型则考虑了相邻残基间的相互关系。
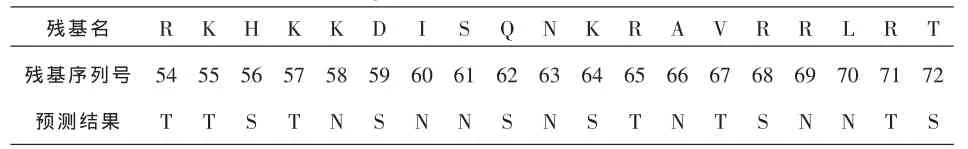
表5 在1xqs_D链上本方法的预测结果
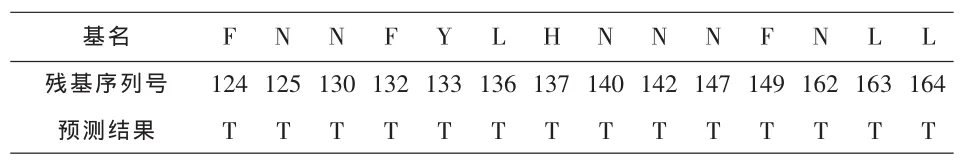
表6 在1fc2_C链上贝叶斯方法的预测结果
5 结论
由实验结果可看出,HMM可以基于已知的一级结构对蛋白质序列进行分类,并进一步预测界面、表面、非表面残基,并且有较好的效果,是对其它分类和预测方法的补充。但它也存在一些缺陷,使得它用于蛋白质分析方面有一定的不足,最主要的因素是Baum-Welch算法存在陷入局部极值、过早收敛或收敛速度慢等缺点,而学习得到的模型参数将直接影响区分和预测算法的准确性。结合HMM 和SVM[7]、贝叶斯[6]、神经网络[8]等方法来改进更好的学习算法,使用更高性能的计算机,结合各种蛋白质结构数据库,将会在蛋白质序列分析方面得到更准确的结果。
[1]张成岗,欧阳曙光,张绍文,等.基于PC/Linux的核酸序列分析系统的构建及其应用[J].生物化学与生物物理进展,2001(2):263-266.
[2]Yona G,Linial N,Linial M.ProtoMap:automatic classification of protein sequences and hierarchy of protein families[J].Nucleic Acids Res,2000,28(1):49-55.
[3]Hideya Kawaji,Yoichi Takenaka,Hideo Matsuda.Graph-based clustering for finding distant relationships in a large set of protein sequences[J].Oxford Journals Life Sciences&Mathematics&Physical Sciences Bioinformatics,2004(20):243-252.
[4]吴晓明,宋长新,王波,等.隐马尔可夫模型用于蛋白质序列分析[J].生物医学工程学杂志,2002,19(3):455-458.
[5]Liu Bin,Homo-complex I data set [DB/OL].(2009-11-20).[2014-02-10].http://www.biomedcentral.com/content/supplementary/1471-2105-10-381-s2.txt.
[6]王池社,程家兴,等.基于贝叶斯方法的蛋白质界面残基预测[J].计算机应用与软件,2009,26(5):75-77.
[7]Qiwen Dong,XiaoLong Wang,Lei Lin,et al.Exploiting residuelevel and profile-level interface propensities for usage in binding sites prediction of proteins[J].BMCBioinformatics,2007(8):147.
[8]Yanay Ofran,Burkhard Rost.ISIS:interaction sites identified fromsequence[J].Bioinformatics,2007(23):2.
[9]欧阳玉梅,方若森.蛋白质-蛋白质界面热点残基预测及其在线工具[J].生命科学,2012,24(1):106-111.
[10]Feihong Wu,Fadi Towfic,Drena dobbs,etc.Analysis of Protein Protein Dimeric Interfaces[C].Fremont:International Conference on Bioinformatics and Biomedicine,2007:35-38.
[责任编辑:桂传友]
TP391
A
1674-1104(2014)03-0025-03
10.13420/j.cnki.jczu.2014.03.007
2014-02-22
池州学院自然科学研究项目(2013ZR017)。
汪一亭(1983-),女,安徽池州人,池州学院数学与计算机科学系助教,硕士,研究方向为生物信息学。
猜你喜欢
生物化学与生物物理进展(2022年6期)2022-07-21
中学生数理化·中考版(2022年6期)2022-06-05
中学生数理化·中考版(2021年6期)2021-11-22
汉字汉语研究(2021年2期)2021-08-30
新世纪智能(数学备考)(2021年4期)2021-08-06
新世纪智能(数学备考)(2021年4期)2021-08-06
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19
汉字汉语研究(2019年2期)2019-08-27
新高考·英语进阶(高二高三)(2018年8期)2018-01-15
河北书画研究(2016年3期)2016-04-28
