
两个天然查尔酮苷的全合成*
2014-08-30 02:12杨金会郭冬冬左武标马晓琴
合成化学 2014年5期
冯 尧,杨金会,郭冬冬,左武标,马晓琴
(宁夏大学 省部共建天然气转化国家重点实验室培育基地,宁夏 银川 750021)
·快递论文·
两个天然查尔酮苷的全合成*
冯 尧,杨金会,郭冬冬,左武标,马晓琴
(宁夏大学 省部共建天然气转化国家重点实验室培育基地,宁夏 银川 750021)
以香草酮和对羟基苯甲醛为原料,经酚羟基保护、羟醛缩合、相转移催化法接糖、去保护基等反应合成了两个天然查尔酮苷——4′-O-β-D-喃葡萄基-3′-甲氧基-4-甲氧基-查尔酮和4′-O-β-D-喃葡萄基-3′-甲氧基-4-羟基-查尔酮,总收率分别为17.2%和20.8%,其结构经1H NMR,13C NMR和HR-MS确证。在脱保护基反应中,首次提出了NH4Cl脱除MOM保护基的新方法。
查尔酮苷;MOM;THP;全合成
黄酮苷类化合物具有广泛的药理活性[1-3],如抗氧化、抗菌、抗病毒等,许多黄酮苷类已作为治疗心血管疾病的药物[4],因此黄酮苷的合成已成为人们关注的热点。
天然黄酮苷在植物中含量很少,而查尔酮苷的化学合成非常罕见。4′-O-β-D-喃葡萄基-3′-甲氧基-4-甲氧基-查尔酮(Ⅰ)和4′-O-β-D-喃葡萄基-3′-甲氧基-4-羟基-查尔酮(Ⅱ)是由Tomohiro Itoh等[5]从芜菁(BrassicarapaL.‘hidabeni’,turnip)中分离到的查尔酮苷。据报道Ⅰ和Ⅱ对大鼠肾上腺嗜铬细胞瘤细胞[6]和正常胶质细胞[7]具有一定抑制活性。因此对Ⅰ和Ⅱ进行全合成研究具有一定的理论意义及潜在的药用前景。
基于本课题组[8-10]对黄酮类化合物的全合成研究基础,本文设计并完成了Ⅰ和Ⅱ的全合成。以对羟基苯甲醛(1)和香草酮(3)为原料,经酚羟基保护、羟醛缩合、相转移催化法接糖、去保护基等反应合成了Ⅰ和Ⅱ(Scheme 1),总收率分别为17.2%和20.8%,其结构经1H NMR,13C NMR和HR-MS确证。在脱保护反应中,首次提出了用NH4Cl脱除MOM保护基的新方法。

1 实验部分
1.1 仪器与试剂
Bruker AM-400MHz型核磁共振仪(CDCl3为溶剂,TMS为内标);FT-IR-8430S型红外光谱仪(KBr压片);Thermo Orbitrap Elite型质谱仪。
2a~2c[12-13]和乙酰溴带葡萄糖(5)[11]按文献方法合成;柱层析用硅胶(200目~300目)和GF254,青岛海洋化工厂;其余所用试剂均为分析纯。
1.2 合成
(1)4a~4c的合成(以4a为例)
在反应瓶中依次加入3552.8mg(3.33mmol),2a453mg(3.33mmol)和乙醇8mL,搅拌使其溶解;冰水浴冷却至0℃,搅拌下缓慢滴加0℃氢氧化钾5.994g(166.5mmol)的水(5mL)和乙醇(6mL)溶液,滴毕,氮气保护反应1h。自然升至室温,于室温反应24h。倒入冰水中,用3mol·L-1盐酸调至pH<3,用乙酸乙酯(3×20mL)萃取,合并有机相,用无水硫酸钠干燥,浓缩后经硅胶柱层析[梯度洗脱剂:V(石油醚)∶V(乙酸乙酯)=20∶1~10∶1]纯化得黄色固体4a576.52mg。
用类似方法合成黄色固体4b和4c。
4′-羟基-3′,4-二甲氧基-查尔酮(4a):产率51.0%;1H NMRδ: 7.79(d,J=15.6Hz,1H,β-H),7.66(d,J=2.0Hz,1H,ArH),7.64(s,1H,ArH),7.61(dd,J=2.0Hz,6.8Hz,2H,ArH),7.44(d,J=15.6Hz,1H,α-H),7.00(d,J=2.4Hz,ArH),6.94(dd,J=2.0Hz,6.8Hz,2H,ArH),3.99(s,3H,CH3),3.88(s,3H,CH3)。
4′-羟基-3′-4-二甲氧甲氧基-查尔酮(4b):产率82%;1H NMRδ: 7.77(d,J=15.6Hz,1H,β-H),7.63(d,J=8.0Hz,1H,ArH),7.61(d,J=1.6Hz,1H,ArH),7.57(dd,J=1.6Hz,8.4Hz,ArH),7.44(d,J=15.6Hz,1H,α-H),7.04(dd,J=1.8Hz,7.0Hz,2H,ArH),6.98(d,J=8.0Hz,1H,ArH),5.19(s,2H,OCH2O),3.90(s,3H,CH3),3.47(s,3H,CH3)。
4′-羟基-3′-甲氧基-4-四氢吡喃基查尔酮(4c):产率63%;1H NMRδ: 7.77(d,J=15.6Hz,1H,β-H),7.64(d,J=2.0Hz,1H,ArH),7.62(dd,J=2.0Hz,3.2Hz,2H,ArH),7.44(d,J=15.6Hz,1H,α-H),7.06(d,J=8.8Hz,2H,ArH),6.98(d,J=8.0Hz,1H,ArH),5.47(t,J=3.0Hz,1H,CH),3.91(s,3H,OCH3),3.86(d,J=1.6Hz,1H,CH),3.62(m,1H,CH),1.90~2.09(m,1H,CH),1.83~1.87(m,2H,CH2),1.52~1.70(m,3H)。
(2)6a~6c的合成(以6a为例)
在圆底烧瓶中加入无水K2CO32.71g(19.6mmol),DMF 35mL和丙酮15mL,搅拌下依次加入4a278mg(0.88mmol),催化量四丁基溴化铵(TBAB)和5761.2mg(2.2mmol),于室温反应13h。旋蒸除去丙酮,残余物用乙酸乙酯(3×40mL)萃取,合并有机相,依次用水(2×5mL)和饱和食盐水(2×5mL)洗涤,无水硫酸镁干燥,旋蒸脱溶后经硅胶柱层析[洗脱剂:A=V(二氯甲烷)∶V(甲醇)=20∶1]纯化得黄色固体6a209mg。
用类似方法合成黄色固体6b和6c。
4′-O-(2,3,4,6-四乙酰基-β-D-吡喃葡萄糖基)-3′-4-二甲氧基-查尔酮(6a):产率38.6%;1H NMRδ: 7.79(d,J=15.6Hz,1H,β-H),7.61~7.54(m,4H,ArH),7.38(d,J=15.6Hz,1H,α-H),7.16(d,J=8.4Hz,1H,ArH),6.93(d,J=8.8Hz,1H,ArH),5.31(dd,J=1.0Hz,3.2Hz,1H,6″-H),5.30(dd,J=0.8Hz,3.2Hz,1H,6″-H),5.18(m,1H,1″-H),5.09(m,1H,5″-H),4.26(dd,J=5.0Hz,12.4Hz,1H,3″-H),4.18(dd,J=2.4Hz,12.4Hz,1H,4″-H),3.89(s,3H,OCH3),3.86(s,3H,OCH3),3.77(m,1H,2″-H),2.07(s,6H,OCH3),2.03(s,6H,OCH3);HR-MSm/z: Calcd for C31H34O13{[M+H]+}637.1892,found 637.1390。
4′-O-(2,3,4,6-四乙酰基-β-D-吡喃葡萄糖基)-3′-4-二甲氧甲氧基查尔酮(6b): 产率35.3%;1H NMRδ: 7.74(d,J=15.6Hz,1H,β-H),7.55(d,J=7.6Hz,2H,ArH),7.53(d,J=8.0Hz,2H,ArH),7.37(d,J=15.6Hz,1H,α-H),7.14(d,J=8.4Hz,1H,ArH),7.03(d,J=8.4Hz,2H,ArH),5.29(dd,J=1.8Hz,3.0Hz,1H,6″-H),5.27(dd,J=3.2Hz,9.2Hz,1H,6″-H),5.15(s,2H,CH2),5.17~5.12(m,1H,1″-H),5.05(dd,J=1.0Hz,3.4Hz,1H,5′-H),4.25(dd,J=5.0Hz,12.2Hz,1H,3″-H),4.15(dd,J=2.2Hz,12.2Hz,4″-H),3.91(s,3H,CH3),3.83~3.79(m,1H,2″-H),3.44(s,3H,CH3),2.05(s,6H,OCH3),2.00(s,6H,OCH3);HR-MSm/z: Calcd for C32H36O14{[M+H]+}667.1997,found 667.1663。
4′-O-(2,3,4,6-四乙酰基-β-D-吡喃葡萄糖基)-3′-甲氧基-4-四氢吡喃基-查尔酮(6c): 产率52.9%;1H NMRδ: 7.73(d,J=15.6Hz,1H,β-H),7.53(d,J=7.6Hz,2H,ArH),7.51(s,2H,ArH),7.37(d,J=15.6Hz,1H,α-H),7.14(d,J=8.8Hz,1H,ArH),7.03(d,J=8.4Hz,2H,ArH),7.29(dd,J=1.9Hz,3.0Hz,1H,6″-H),7.26(d,J=2.8Hz,1H,6″-H),5.29(dd,J=1.8Hz,3.0Hz,6″-H),5.17(s,2H,OCH2O),5.13(t,J=2.5Hz,1H,1″-H),5.06(dd,J=1.0Hz,3.2Hz,1H,5″-H),4.25(dd,J=5.0Hz,12.2Hz,1H,3″-H),4.15(dd,J=2.2Hz,12.2Hz,1H,4″-H),3.86(s,3H,OCH3),3.83~3.80(m,1H,2″-H),3.44(s,3H,OCH3),2.04(s,6H,COCH3),2.00(s,6H,COCH3);HR-MSm/z: Calcd for C35H40O14{[M+H]+}707.2310,found 707.2305。
(3)Ⅰ的合成
在圆底烧瓶中加入6a15mg(0.024mmol)和甲醇5mL,搅拌下滴加NH3·H2O 1.5mL,滴毕,于室温反应30min。旋蒸除溶后经硅胶柱层析(洗脱剂:A=5∶1)纯化得黄色粉末Ⅰ 10mg,产率92%;1H NMR(MeOD)δ: 7.77(dd,J=2.0Hz,8.8Hz,1H,6′-H),7.72(d,J=15.6Hz,1H,β-H),7.71(dd,J=2.0Hz,7.2Hz,2H,2,6-H),7.62(d,J=2.0Hz,1H,2′-H),7.64(d,J=15.6Hz,1H,α-H),7.27(d,J=8.4Hz,1H,5′-H),6.99(dd,J=1.8Hz,6.8Hz,2H,3,5-H),5.07(d,J=7.6Hz,1H,1″-H),3.94(s,3H,OCH3),3.88(d,J=2.0Hz,6″-H),3.89(s,3H,OCH3),3.74(d,J=14.8Hz,1H,6″-H),3.54(t,J=6.8Hz,1H,2″-H),3.48~3.50(m,2H,3″,5″-H),3.42(t,J=8.8Hz,1H,4″-H);13C NMR(MeOD)δ: 188.8,161.5,150.3,148.8,143.9,132.0,130.6,129.7,127.0,122.3,118.1,114.2,113.5,112.7,111.1,110.8,99.9,76.4,75.9,72.8,69.3,60.5,54.7,54.0;HR-MSm/z: Calcd for C32H35O14{[M+H]+}469.1469,found 469.1467。
(4)Ⅱ的合成
在圆底烧瓶中加入6b34mg(0.053mmol)和甲醇10mL,搅拌下滴加NH3·H2O 3mL,滴毕,反应0.5h(TLC检测)。缓慢滴加3mol·L-1盐酸调至pH6,于70℃反应1h。旋蒸除溶得黄色固体Ⅱ 17.7mg,产率77.3%;1H NMR(MeOD)δ: 7.77(dd,J=1.8Hz,6.6Hz,1H,6′-H),7.74(d,J=11.6Hz,1H,β-H),7.66(d,J=1.2Hz,1H,2′-H),7.63(d,J=6.0Hz,2H,2,6-H),7.60(d,J=15.6Hz,1H,α-H),7.27(d,J=8.4Hz,1H,5′-H),6.85(dd,J=1.4Hz,8.6Hz,2H,3,5-H),8.07(d,J=7.2Hz,1H,1″-H),3.94(s,3H,OCH3),3.91(dd,J=2.0Hz,12.0Hz,1H,6″-H),3.71(dd,J=5.3Hz,12.2Hz,1H,6″-H),3.54(t,J=7.6Hz,1H,2″-H),3.48~3.51(m,2H,3″,5″-H),3.42(t,J=8.4Hz,1H,4″-H);13C NMR(MeOD)δ: 189.0,159.7,151.3,150.3,148.6,47.3,144.4,143.8,132.1,129.9,129.8,129.7,125.9,125.8,123.0,122.2,117.4,115.0,114.2,113.9,110.9,110.4,99.9,76.4,75.9,72.8,69.3,60.5,54.7,28.8,20.1,18.6;HR-MSm/z: Calcd for C22H24O9{[M+H]+}455.1313,found 455.1310。
(5)4′-O-(2,3,4,6-四乙酰基-β-D-吡喃葡萄糖基)-3′-甲氧基-4-羟基-查尔酮(Ⅲ)的合成[13]
在反应瓶中依次加入6c110mg(0.16mmol)和甲醇10mL,搅拌使其溶解;加入催化量浓硫酸改性活性炭,于室温反应30min(TLC检测)。过滤除去浓硫酸改性活性炭,滤液旋干溶剂后经硅胶柱层析(洗脱剂:A=5∶1)纯化得黄色固体Ⅲ 79mg,产率79.2%;1H NMRδ: 7.74(d,J=15.6Hz,1H,β-H),7.55(d,J=9.2Hz,4H,ArH),7.37(d,J=15.6Hz,1H,α-H),7.14(d,J=8.4Hz,1H,ArH),7.05(d,J=8.8Hz,2H,ArH),5.45(t,J=3.0Hz,1H,CH),5.29(dd,J=1.8Hz,2.8Hz,2H,6″-H),5.15(m,1H,1″-H),5.06(dd,J=1.2Hz,7.6Hz,1H,5″-H),4.24(d,J=4.8Hz,1H,3″-H),4.17(dd,J=2.4Hz,9.6Hz,1H,4″-H),3.86(s,3H,OCH3),3.84~3.79(m,2H,CH2),3.60~3.55(m,1H,CH),2.05(s,6H,COCH3),2.01(s,6H,COCH3),1.82~1.85(m,2H,CH2),1.50~1.70(m,3H)。
2 结果与讨论
2.1 合成
(1)Ⅰ的合成
以对羟基苯甲醛为原料,与硫酸二甲酯在丙酮中反应得2a;2a与3经羟醛缩合得查尔酮4a;4a通过相转移催化接糖的方法以38.6%的产率制得6a;6a在NH3·H2O作用下脱去乙酰基得Ⅰ。
(2)Ⅱ的合成
2011年Hirokazu Hara等[5]采用乙酰基保护酚羟基,Ag2CO3/pyridine接糖,最后脱去乙酰基的方法完成了Ⅱ的合成;2013年李苗苗等[12]采用苄基保护羟基,相转移催化的方法接糖,最后脱去苄基以及乙酰基合成了Ⅱ。
本文分别采用MOM和THP两种羟基保护基策略,用相转移催化的方法接糖,最后脱去MOM/THP以及乙酰基,完成了Ⅱ的合成。
MOM保护羟基的合成路线:4b经TBAB相转移催化法接糖,然后一步脱除乙酰基以及MOM保护基合成Ⅱ。值得一提的是6b在脱除保护基过程中,我们采用一锅法操作同时脱除了Ac和MOM保护基,直接得到了目标化合物Ⅱ。首先,用NH3·H2O脱去乙酰基,反应结束后,滴加3mol·L-1盐酸中和NH3·H2O至弱酸性(pH≈6),于70℃反应约1h,随后蒸除去溶剂,分离出有机物,即为Ⅱ。该方法直接脱除乙酰基以及MOM保护基,且最终产率较高(77.3%)。
我们还尝试了用THP保护羟基合成Ⅱ。用相转移催化法接糖时,有THP脱除产物6c和含有THP的化合物Ⅲ生成。这种方法接糖的产率较MOM、乙酰基、苄基等保护基高,总产率几乎达100%。脱去THP时首先尝试用蒙脱土K10,但未获得产物。最后用硫酸改性活性炭以79.2%的产率脱去了THP[13]。Ⅲ脱去乙酰基得Ⅱ。
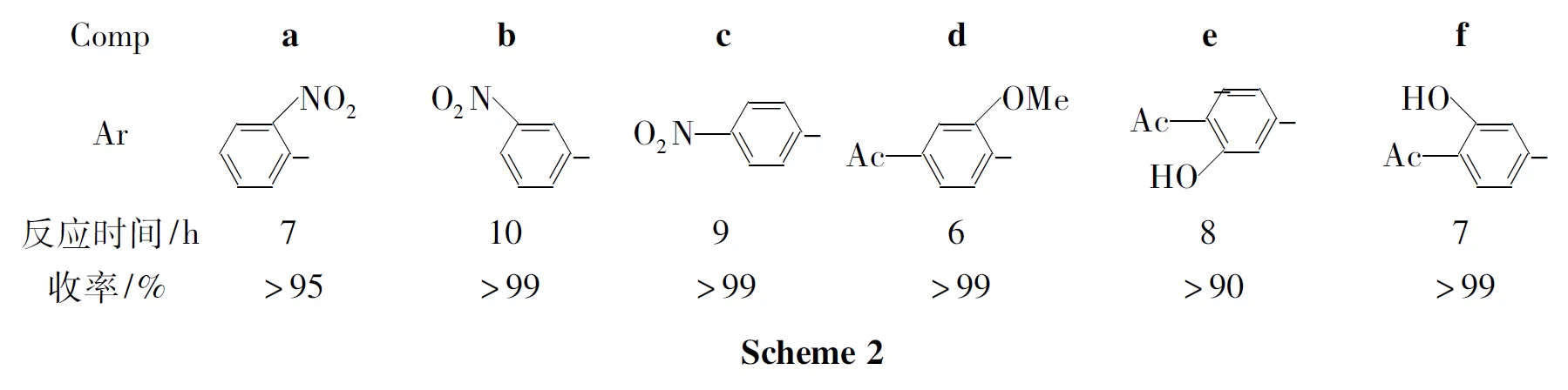
2.2 脱去MOM的新方法
MOM保护基为较为稳定的羟基保护基,需要盐酸、对甲基苯磺酸等较强酸脱除,而这类强酸会破坏糖苷键。本文报道一种脱去MOM的新方法:采用强酸弱碱盐NH4Cl在甲醇/水中加热脱去MOM。并尝试采用该方法对几种化合物(Ar-MOM,Scheme 2)脱去MOM,发现NH4Cl均能较好的脱去MOM保护基,但反应时间较长。脱去MOM保护基与溶液中H+的浓度有关,溶液中水的比例越大,NH4Cl的溶解度越大,H+的浓度越大,MOM保护基脱去越快。但一般有机物水溶性较差,影响溶液中H+的浓度,因此NH4Cl脱MOM的方法对于水溶性较好的化合物如糖类较为适合。
3 结论
采用温和有效的方法完成了两个查尔酮苷(Ⅰ和Ⅱ)的合成,为该类化合物的合成提供了参考。提出了脱去MOM的新方法,为有机合成中保护基的使用和脱去提供了新的思路。
[1] Harbone J B,Williams C A.Advances in flavonoid research since 1992[J].Phytochemistry,2000,55:481-504.
[2] Pietta P G.Flavonoids as antioxidants[J].J Nat Prod,2000,63(7):1035-1042.
[3] Harborne J B,Baxter H.The Handbook of Natural Flavonoids[M].New York:John Wiley &Sons Chichester,1999.
[4] Costas D,Alexios L,Franç T,etal.Phase-transfer catalyzed synthesis of flavonoid glycosides[J].Carbohydr Res,1990,207:131-137.
[5] Masayuki Ninomiya,Mamoru Koketsu,Toshiyasu I,etal.Chalcone glycosides from aerial parts of Brassica rapa L.‘hidabeni’,turnip[J].Phytochemistry Letters,2010,3:96-99.
[6] Atsuyoshi Nishina,Hirokazu Kimura,Hiroyuki T,etal.Neurite outgrowth of PC12Cells by 4′-O-β-D-glucopyranosyl-3′,4-dimethoxychalcone from Brassica rapa L.‘hidabeni’ was enhanced by pretreatment with p38MAPK inhibitor[J].Neurochem Res,2013,38:2397-2407.
[7] Hirokazu Haraa,Yoko Nakamura,Masayuki N,etal.Inhibitory effects of chalcone glycosides isolated from Brassica rapa L.‘hidabeni’ and their synthetic derivatives on LPS-induced NO production in microglia[J].Bioorganic &Medicinal Chemistry,2011,19:5559-5568.
[8] 杨金会,谢一民,冯尚彪,等.四种天然双异戊烯基黄酮的合成及其抑菌活性研究[J].有机化学,2013,32:1-8.
[9] 左武标,杨金会,李红俊,等.四个天然异戊烯基黄酮的全合成研究[J].有机化学,2012,32:1-7.
[10] 郭冬冬,杨金会,梁西周,等.两种天然异戊烯基黄烷酮的全合成[J].合成化学,2013,21(5):550-553.
[11] 凌勋利.糖基给体溴代糖的合成[J].洛阳师范学院学报,2008,2:75-77.
[12] 李苗苗.查尔酮糖苷类化合物的全合成研究[D].兰州:兰州理工大学,2012.
[13] 张萱,杨金会,江世智,等.改性活性炭催化羟基的四氢吡喃化保护及脱保护反应研究[J].合成化学,2008,16(6):632-635.
TotalSynthesisofTwoNaturalChalconeGlycosides
FENG Yao, YANG Jin-hui, GUO Dong-dong, ZUO Wu-biao, MA Xiao-qin
(State Key Laboratory Cultivation Base of Natural Gas Conversion,Ningxia University,Yinchuan 750021,China)
Two natural chalcone glycosides,4′-O-β-D-glucopyranosyl-3′,4-dimethoxychalcone and 4′-O-β-D-glucopyranosyl-4-hydroxy-3′-methoxychalcone,were synthesized by protection of phenolic hydroxyl group,aldol condensation,glycosylation under phase transfer catalytic condition and deprotection starting from acetovanillone,and the total yield were 17.2% and 20.8%.The structures were confirmed by1H NMR,13C NMR and HR-MS.A new method for removing MOM group with NH4Cl was also provided.
chalcone glycoside;MOM;THP;total synthesis
2014-05-21
国家自然科学基金资助项目(21362025,21162021);教育部“新世纪优秀人才支持计划资助”(NCET-09-0860);宁夏自然科学基金资助项目(NZ1006)
冯尧(1989-),男,汉族,宁夏中卫人,硕士研究生,主要从事天然产物的全合成研究。
杨金会,博士,教授,Tel.0951-2062246,E-mail: yang_jh@nxu.edu.cn
O629.1;O621.3
A
1005-1511(2014)05-0638-05
猜你喜欢
陕西师范大学学报(自然科学版)(2022年5期)2022-11-09
化学工程师(2022年5期)2022-05-11
云南化工(2020年4期)2020-05-19
科海故事博览·中旬刊(2020年3期)2020-03-15
安徽化工(2018年5期)2018-10-23
天然产物研究与开发(2018年9期)2018-10-08
小猕猴学习画刊(2016年9期)2016-05-14
合成化学(2015年9期)2016-01-17
烟草科技(2015年8期)2015-12-20
影像科学与光化学(2014年3期)2014-03-11
