
CryⅢA基因植物表达载体构建及马铃薯遗传转化
2014-11-08 03:07尤佳张宁文义凯吴家和司怀军王蒂
草业学报 2014年1期
尤佳,张宁,文义凯,吴家和,司怀军*,王蒂
(1.甘肃省干旱生境作物学省部共建国家重点实验室培育基地 甘肃省作物遗传改良与种质创新重点实验室,甘肃 兰州 730070;2.甘肃农业大学生命科学技术学院,甘肃 兰州 730070;3.中国科学院微生物研究所植物基因组学国家重点实验室,北京 100101)
马铃薯甲虫(Leptinotarsadecemlineata)属鞘翅目叶甲科,又称马铃薯叶甲或科罗拉多马铃薯甲虫(Colorado potato beetle, CPB),是国际公认的毁灭性检疫害虫,也是我国重要外来入侵物种之一。因其具有高繁殖率、滞育和迁飞等习性,生态可塑性强,且世代重叠,防治难度大,其主要以成虫和幼虫取食叶片,通常将植株叶片吃光,仅剩茎秆,其危害造成马铃薯产量一般损失为30%~50%,严重者可达90%以上,甚至绝产。而且还可传播病害,如褐斑病、环腐病等[1-2]。近年来我国检疫部门通过口岸检疫截获马铃薯甲虫的频率在逐步增加[3]。由此可见,我国马铃薯甲虫的防控形势十分严峻。利用植物基因工程的方法选育优良的抗虫植物品种已被证实为一种控制虫害的有效途径[4]。目前全世界总共已获得了50多种转Bt基因植物[5],在已获得的转Bt基因植物中,绝大多数植物对靶标昆虫产生了强烈的毒杀作用,有效地减轻了由害虫所造成损失。尽管转Bt基因抗虫植物在生产上已经展现出了良好的应用前景,但依旧存在一些问题需要妥善地加以解决,否则就有可能影响其持续利用,甚至产生一些不良后果,其中昆虫对转Bt杀虫晶体蛋白产生抗性的问题尤为严峻[6]。Bt-CryⅢA基因是一种能抗鞘翅目(Coleoptera)昆虫的基因,Sutton等[7]对特异毒杀鞘翅目昆虫的CryⅢA基因进行了重新合成。试验显示,表达该基因的烟草(Nicotianatabacum)蛋白质提取物对CPB有明显的抑制作用,表明改造后的CryⅢA基因在抗甲虫转基因植物研究上具有应用价值,对抑制检疫性害虫CPB具有一定作用。Perlak等[8]用合成的CryⅢA基因,也获得了可有效抵抗CPB的转基因马铃薯(Solanumtuberosum)植株。1995年,孟山都公司转CryⅢA基因的抗甲虫马铃薯新品种(Newleaf)获准商业化生产。
本研究选用的CryⅢA基因内部含有植物表达载体pBI121的酶切位点,选用传统的酶切连接方法需要采用平端连接的方法,为此首先需要Klenow酶将酶切后突出的5′末端填平,再用T4 DNA聚合酶将突出的3′末端删去。为了减少载体的自身环化,还经常采用牛小肠碱性磷酸酶将载体5′末端的磷酸基团去除[9]。但平端连接的效率非常低,而且较难确定插入片段的正反方向。为了实现定向克隆,人们常采用TA克隆载体进行重组,但要将目的基因插入片段克隆到所需要的载体上尚需一轮亚克隆,因此操作比较繁琐,实验难度大,工作周期长[10]。
In-Fusion克隆技术是新兴发展起来的克隆技术,该技术的原理是目的基因的PCR引物的5′端加上与线性化载体同源的碱基序列,以使PCR的扩增产物两端分别带上15个和线性化载体两端同源的碱基序列。In-Fusion酶可以识别PCR产物和线性化载体两端的同源序列,将PCR产物置换到目标载体上从而发生同源重组,这种技术不受载体和酶切位点限制,不附加冗余序列,适用于各种载体模型构建,也可以完成多片段连接实验和定点突变实验,是一种快捷高效的克隆技术。本研究采用In-Fusion克隆技术成功构建了组成型启动子CaMV 35S和马铃薯茎叶特异表达启动子ST-LS1驱动的CryⅢA基因植物表达载体,并转化马铃薯获得了转基因植株,对CryⅢA基因在转基因植株中的表达特性进行了分析,为选育抗甲虫转基因马铃薯新品系奠定了一定的基础。
1 材料与方法
1.1 材料
1.1.1菌株和载体 大肠杆菌(Escherichiacoli)DH5α、根癌农杆菌(Agrobacteriumtumefaciens)LBA4404及植物表达载体pBI121由甘肃农业大学作物遗传改良与种质创新重点实验室提供。含合成的苏云金芽孢杆菌杀虫晶体蛋白CryⅢA基因的质粒pBin438-CryⅢA[11],由中国科学院微生物研究所吴家和副研究员提供。将pBI121中的组成型启动子CaMV 35S置换为马铃薯茎叶特异表达启动子ST-LS1的质粒的pBI121-ST-LS1由本实验室构建并保存。
1.1.2酶及生化试剂 LATaqDNA 聚合酶,限制性核酸内切酶HindⅢ、BamHⅠ、SacⅠ,PrimeScript RT reagent Kit with gDNA Eraser,SYBR®Premix Ex TaqTMGC购自TaKaRa公司;In-Fusion®HD Cloning Kit购自Clontech公司;DNA胶回收试剂盒购自上海生物工程技术服务有限公司。常用的化学试剂为国产分析纯。引物由上海生物工程技术服务有限公司合成。溶液配方及常用抗生素与激素的配置详见文献[12]。
1.1.3引物设计 根据已知的CryⅢAcDNA的序列设计合成扩增引物,为了使扩增的目的片段与经BamHⅠ和SacⅠ酶切后的线性化pBI121载体发生同源重组反应,在扩增目的片段的上、下游引物的5′端分别加入与载体线性化位点两侧序列同源的15~20 bp。下划线处为鉴定酶切位点,虚线为根据CryⅢA的CDS区序列设计的引物。正向引物infusion-F1: 5′-GGACTCTAGAGGATCCATGACTGCTGATAACAACACG-3′(下划线为BamHⅠ位点)和反向引物infusion-R1:5′-GATCGGGGAAATTCGAGCTCTTAATTCACTGGAATGAA CTC-3′(下划线为SacⅠ位点)。同理,准确找出茎叶特异表达启动子ST-LS1启动GUS的表达载体pBI121-ST-LS1被限制性内切酶BamHⅠ、SacⅠ酶切成为线性载体后两端15~20 bp的准确序列,与根据CryⅢA的CDS区序列设计的引物按照要连接的方向拼成完整的定制引物。正向引物infusion-F2:5′-GTGAGAGCAAGGATCCATGACTGCTGATAACAACACG-3′(下划线为BamHⅠ位点)和反向引物infusion-R2: 5′-GATCGGGGAAATTCGAGCTCTTAATTCACTGGAATGAACTC-3′(下划线为SacⅠ位点)。另外在CryⅢA的CDS区序列中间设计一对普通引物作为检测引物,序列为JCP-F:5′-CAGAAGATTGCCGATTACG-3′和JCP-R:5′-GAGTCGTTACCGTAGTATCCTG-3′,扩增片段大小为709 bp。引物由上海生物工程技术服务有限公司合成,用无菌去离子水溶解至终浓度为10 μmol/L,-20℃保存备用。
1.2 植物表达载体的构建
1.2.1目的片段的PCR 提取pBin438-CryⅢA的质粒为模板,分别用正向引物infushion-F1、反向引物infusion-R1和正向引物infusion-F2、反向引物infusion-R2进行PCR扩增,反应程序:94℃ 4 min, 94℃ 1 min,55℃ 1 min 30 s,72℃ 1 min,35个循环,72℃ 10 min,4℃保存。用1.5%琼脂糖进行PCR产物凝胶电泳,用DNA快速纯化回收试剂盒回收目的片段,具体操作按产品说明书进行。
1.2.2植物表达载体pBI121线性化处理BamHⅠ和SacⅠ限制性酶双酶切植物表达载体pBI121和pBI121-ST-LS1。酶切反应体系为:质粒DNA 8 μL+ddH2O 8 μL+10×K Buffer 2 μL+BamHⅠ1 μL+SacⅠ1 μL,反应体系共20 μL。37℃恒温酶切16 h,1.5%的琼脂糖凝胶电泳检测酶切结果,从胶上切下目的条带,用DNA快速纯化回收试剂盒回收目的片段,具体操作按产品说明书进行。
1.2.3建立In-Fusion连接体系 回收后的线性化载体和目的基因用Implen超微量分光光度计测浓度,根据目的基因与载体摩尔比10∶1来计算出各所需的体积,连接体系如下:5× In-Fusion HD Enzyme Premix 2 μL+Linearized Vector 6 μL+Purified PCR Fragment 2 μL,反应体系共10 μL。50℃水浴反应15 min,-20℃贮存。组成型启动子CaMV 35S和马铃薯茎叶特异表达启动子ST-LS1驱动的CryⅢA基因植物表达载体pBI121-CaMV35S-CryIIIA和pBI121-ST-LS1-CryIIIA构建示意图分别见图1和图2。
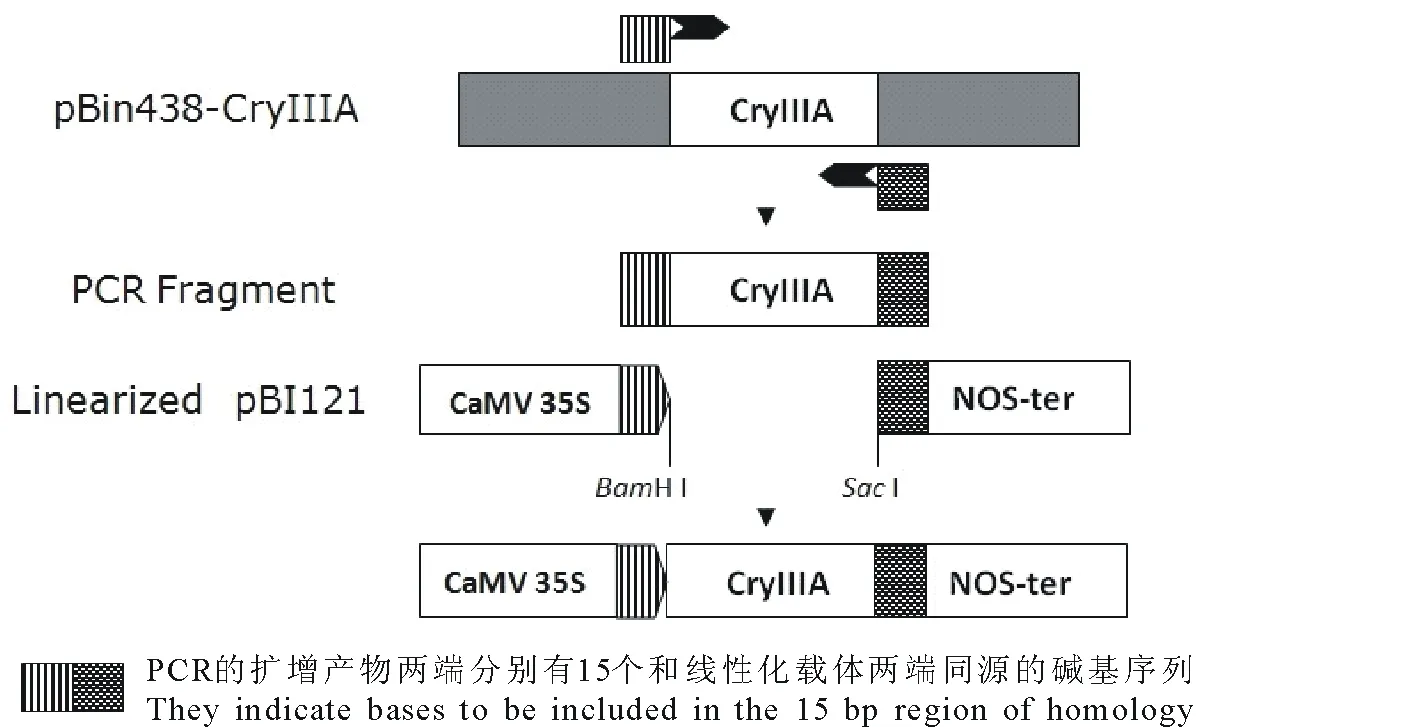
图1 植物表达载体pBI121-CaMV35S-CryIIIA构建示意图
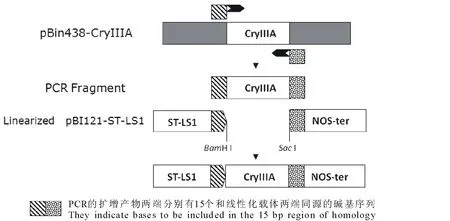
图2 植物表达载体pBI121-ST-LS1-CryIIIA构建示意图
将上述连接产物转化大肠杆菌 DH5α感受态细胞,用质粒PCR和酶切鉴定的方法证明构建是否成功。CaMV 35S驱动的CryⅢA基因重组载体质粒用BamHⅠ和SacⅠ进行双酶切鉴定;ST-LS1驱动CryⅢA基因重组载体除了用BamHⅠ和SacⅠ进行双酶切鉴定外,用BamHⅠ和HindⅢ也进行一次双酶切。双酶切反应体系:质粒DNA 8 μL+ddH2O 8 μL+10×K Buffer 2 μL+BamHⅠ1 μL+SacⅠ(HindⅢ)1 μL,37℃,6 h。1%的琼脂糖凝胶电泳检测酶切结果。目的基因序列的测定由上海生工有限公司完成。正确后即可用于农杆菌的转化。农杆菌的转化方法见文献[13],转化及鉴定方法参照文献[14-17]。
1.3 农杆菌介导的马铃薯试管薯遗传转化与植株再生
1.3.1马铃薯试管薯的诱导 以马铃薯栽培品种陇薯3号和甘农薯2号为实验材料。试管苗在无菌条件下剪成带有2个腋芽的茎段,转接到MS+3%蔗糖+0.45%琼脂的固体培养基上。用150 mL三角瓶培养,每瓶装50 mL固体培养基,接种5个茎段,2000 lx光照下培养3 周,形成壮苗。将试管苗在MS+3%蔗糖的液体培养基上生长3 周后,倒出三角瓶中的液体培养基,加入50 mL的诱导培养基[MS+5 mg/L 6-BA(6-苄氨基嘌呤,6-benzylaminopurine)+8%蔗糖],置于暗处条件下诱导结薯。
1.3.2诱导芽与生根选择压的确定 将直径为5 mm左右的马铃薯试管薯从三角瓶里取出后切成厚度为1 mm左右的薄片,分别接种于附加 0,25,50,75 mg/L Kan(卡那霉素,kanamycin)的芽诱导培养基上,培养基为 MS+1 mg/L IAA(吲哚-3-乙酸,indole-3-acetic acid)+0.2 mg/L GA3(赤霉素,gibberellic acid)+0.5 mg/L 6-BA+2 mg/L ZT(玉米素核苷,zeatin riboside)。培养4 周后,确定抑制芽分化的最低 Kan浓度。将无菌试管苗剪成带有2个腋芽的茎段接种于分别附加 50,75,100 mg/L Kan的MS培养基上,3 周后观察无菌苗的生根状况,并确定抑制生根的最低Kan浓度。
1.3.3农杆菌工程菌液的制备 吸取1 mL含目的基因的农杆菌菌液接种于50 mL含Kan(50 mg/L)和Rif(利福平,rifampicin,50 mg/L)的LB液体培养基中,于28℃、260 r/min避光振荡培养过夜。吸取1 mL接种于50 mL含 Kan(50 mg/L)和Rif(50 mg/L)的LB液体培养基中28℃、260 r/min避光振荡培养3~4 h至对数生长期,然后转入无菌离心管中,于5000 r/min离心6 min,收集菌体用等量液体MS培养基重新悬浮后用于转化,其处理所用菌液浓度OD600为0.5。
1.3.4外植体侵染与植株再生 将直径为0.5 cm左右的试管薯切成2 mm的薄片,置于农杆菌工程菌液中浸泡7 min,期间不断摇动,使菌液与薯片充分接触。然后用无菌滤纸吸去多余菌液,转入固体MS培养基于28℃、黑暗条件下共培养2 d,然后转移到分化培养基[MS+1 mg/L IAA+0.2 mg/L GA3+0.5 mg/L 6-BA+2 mg/L ZT+50 mg/L Kan+200 mg/L Carb(羧苄青霉素,carbenicillin)],置于光照度2000 lx,光周期16 h/d,温度24℃的条件下,10 d换一次培养基,培养3~4 周即可分化出芽。待芽长0.5~1.0 cm时切下转入生根培养基(MS+100 mg/L Kan+200 mg/L Carb),进行生根筛选和扩繁。
1.4 转基因植株鉴定
1.4.1转化再生植株的 PCR检测 将待检测植株和对照植株的叶片用 CTAB(十六烷基三甲基溴化胺,cetyltrimethyl ammonium bromide)法提取总DNA。参照 Sambrook等[14]的方法对抗性植株进行 PCR 检测。以检测引物JCP-F和JCP-R进行 PCR 扩增,预期片段大小为709 bp。用1%琼脂糖凝胶电泳检测 PCR 结果。
1.4.2转基因植株的实时荧光定量(qRT-PCR)检测与分析 分别提取DNA检测呈阳性植株的茎、叶和根总RNA,用PrimeScript RT reagent Kit with gDNA Eraser反转录试剂进行反转录,具体操作按产品说明书进行。以马铃薯ef1a基因为内参(扩增引物为ef1a-F:5′-CAAGGATGACCCAGCCAAG-3′和ef1a-R:5′-TTCCTTACCTGAACGCCTGT-3′),以CryⅢA基因序列设计一对特异性较高的引物Bt-F:5′-TCTACAGAGCCGTGGCAAAC-3′和Bt-R:5′-CTGGGATGGTTC CTCTGCTAC-3′。利用SYBR荧光染料法进行转基因植株实时荧光定量(qRT-PCR)检测与分析[14],根据公式2-△△Ct计算出CryⅢA基因在根、茎、叶中的相对表达量。
2 结果与分析
2.1 植物表达载体构建
2.1.1组成型表达启动子CaMV 35S驱动的CryⅢA基因表达载体构建 对重组载体进行质粒PCR鉴定,检测引物JCP-F和JCP-R扩出约700 bp大小的条带(图3泳道1),引物infusion-F1和infusion-R1扩出约1800 bp大小的条带(CryⅢA基因大小为1794 bp,引物还含有线性化载体两端共36 bp大小的碱基),扩增得到的目的片段与预期大小一致(图3泳道2)。用BamHⅠ和SacⅠ进行重组质粒双酶切鉴定,获得大小约13000,1500,300 bp三条带(CryⅢA基因第328个碱基位置含有SacⅠ的酶切位点,用SacⅠ进行酶切不仅可以将其从重组载体上切下,而且会从中间切断,基因整体大小为1794 bp,酶切后会成为1466和328 bp两个片段),得到目的片段大小均与预期大小一致(图4)。 重组质粒经上海生工有限公司测序表明得到的序列正确。
2.1.2马铃薯茎叶特异表达启动子ST-LS1驱动的CryⅢA基因表达载体构建 对重组载体进行质粒PCR鉴定,检测引物JCP-F和JCP-R扩出约700 bp大小的条带(图5泳道2),引物infusion-F2和infusion-R2扩出约1800 bp大小的条带,与预期大小一致(图5泳道1)。分别用BamHⅠ和SacⅠ、BamHⅠ和HindⅢ进行重组质粒双酶切鉴定,BamHⅠ和SacⅠ切出大小应为13000,1500和300 bp三条带(CryⅢA基因第328个碱基位置含有SacⅠ的酶切位点,用SacⅠ进行酶切后会成为1466和328 bp两个片段)。BamHⅠ和HindⅢ双酶切得到大小约14000和1600 bp两条带(茎叶特异启动子ST-LS1启动GUS的表达载体pBI121大小为15459 bp,切掉GUS连上CryⅢA基因大小为15359 bp,启动子大小为1572 bp),得到目的片段大小均与预期大小一致(图6)。重组质粒经上海生工有限公司测序表明得到的序列正确。
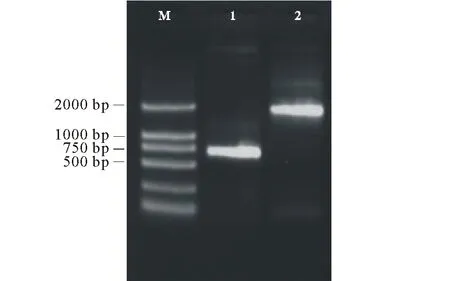
图3 重组质粒pBI121-CaMV35S-CryIIIA的PCR扩增鉴定
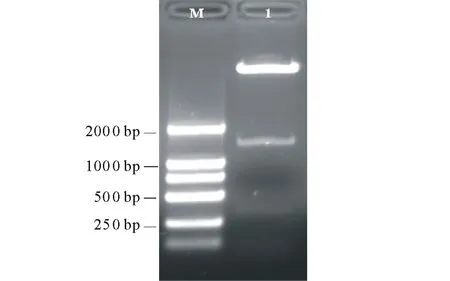
图4 重组质粒pBI121-CaMV35S-CryIIIA的酶切鉴定
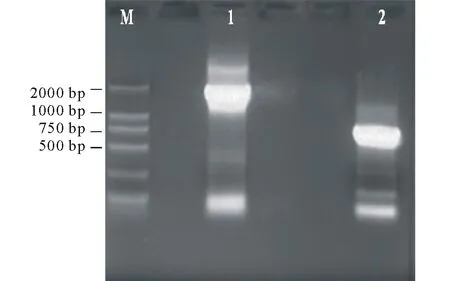
图5 重组质粒pBI121-ST-LS1-CryIIIA的PCR扩增鉴定
利用冻融法将2个重组质粒转入根癌农杆菌LBA4404,挑取单克隆的转化菌接种培养,提取质粒DNA进行PCR鉴定,能特异地扩增出大小约1800 bp(引物infusion-F2和infusion-R2)和700 bp(引物JCP-F和JCP-R)左右的片段,表明2个重组质粒均成功转入农杆菌中。
2.2 马铃薯遗传转化与生根选择压的确定
马铃薯试管薯薄片在芽分化培养基上经过2~3 周的培养,未转化对照组的外植体能直接分化长出绿色健壮小芽;在25 mg/L Kan选择浓度下,农杆菌转化外植体分化芽的情况与对照无明显差异;在50 mg/L Kan选择浓度下,对照组外植体已经基本不能分化出芽。因而确定芽诱导时Kan的选择压为50 mg/L。4 周后,转化试管薯薄片分化出绿色小芽(图7)。马铃薯植株经过3 周的生根选择培养,未转化对照组的植株未能生根(图8),而转化植株能够正常生根(图8,9)。
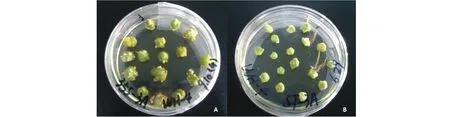
图7 试管薯薄片在含有卡那霉素的分化培养基上再生出绿色小芽

图8 转化植株在附加100 mg/L Kan培养基上进行生根选择

图9 转化植株在附加100 mg/L Kan培养基上生根
2.3 转基因植株检测与分析
PCR扩增产物的电泳分析结果表明,转化植株可扩增到709 bp的特异性条带,而未转化的植株中无该片段的产生(图10),从而初步证明再生植株为转基因植株。经Kan抗性生根筛选和PCR扩增鉴定共获得10株抗性植株,对其进行qRT-PCR检测,结果表明,转基因马铃薯植株有典型的荧光扩增曲线;且其Ct值都小于40,而非转基因植株和空白对照其荧光扩增曲线都呈现平直,且都位于阈值线之下。CaMV 35S为组成型强启动子,其驱动的CryⅢA转基因马铃薯在不同组织中基因均有表达,从相对表达量来看,CryⅢA基因在马铃薯根、茎、叶中的表达有显著差异,其中叶的平均表达量是茎和根的4~5倍(图11);茎叶特异表达启动子ST-LS1驱动的CryⅢA转基因马铃薯茎、叶中有表达,根的荧光扩增曲线呈现平直,未见表达,从相对表达量来看,叶的表达量也高出茎4倍左右(图12)。
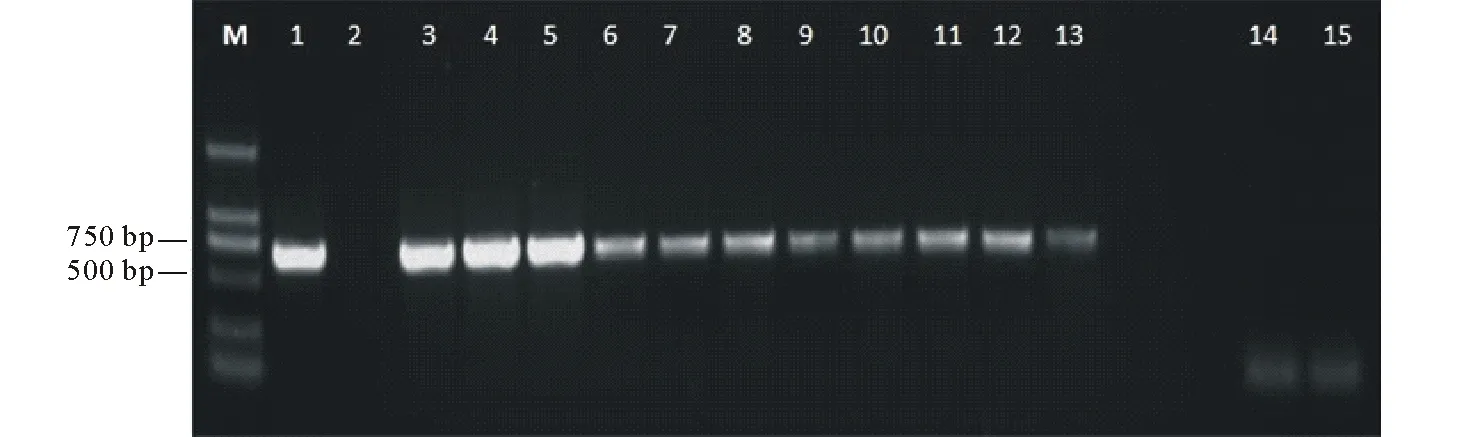
图10 部分转基因植株的PCR检测
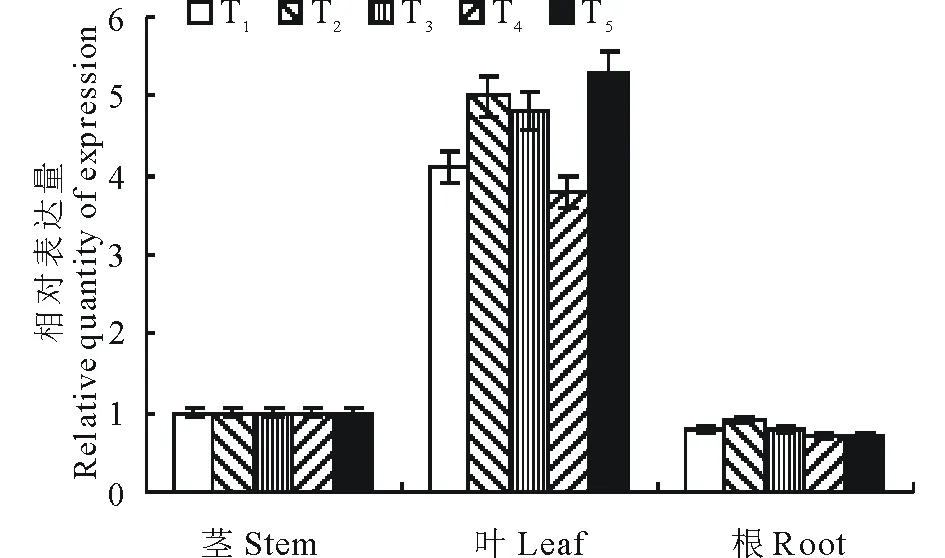
图11 pBI121-CaMV35S-CryIIIA转基因植株的qRT-PCR组织特异性表达检测

图12 pBI121-ST-LS1-CryIIIA转基因植株的qRT-PCR组织特异性表达检测
3 讨论
基因重组技术是分子生物学中常用的方法,是研究基因功能的基本手段。但在基因重组中经常会出现插入的目的基因片段与载体的酶切位点不匹配,致使基因重组出现困难。为解决酶切位点不匹配的情况,人们常采用平端连接,但是平端连接的操作步骤多,连接效率极低以及需要鉴定正反向插入等缺点。为克服上述困难,本实验采用In-Fusion技术成功构建了组成型启动子CaMV 35S和马铃薯茎叶特异表达启动子ST-LS1驱动的CryⅢA基因植物表达载体。In-Fusion技术构建重组质粒具有以下优点:适用范围极广,平、粘末端连接都适用,只要线性化载体就行;不用连接酶、不用载体的去磷酸化、反应混合物50℃水浴15 min即可,节省了时间;可以实现靶向克隆,避免了鉴定正反向插入的麻烦;插入的目的基因从起始密码子序列开始,没有两端多余的序列,不影响后续基因的表达;重组效率高;无需末端抹平。该方法避免了传统克隆方法难以克服的困难,是一种简便高效、省时省力的基因克隆手段。实验能否成功的关键在于引物的设计。这种方法引物设计的原则是引物的5′末端必须包含与载体末端相同的15个碱基序列,引物的3′末端必须包含与目的基因片段相互补的特异碱基序列,补齐酶切位点缺失碱基。该实验采用的infusion酶,可以识别PCR产物和线性化载体的两端同源序列,并将PCR产物置换到目标载体上从而发生同源重组。在这一过程中,为了提高同源重组率,目的基因片段和目标载体的比例要合适,二者的摩尔数之比一般(8~10)∶1较好。PCR产物浓度会比酶切回收的载体浓度高很多,所以合理控制二者比例才能有效连接。
CryⅢA基因编码的Bt毒素蛋白具有对人、畜及害虫天敌极少或完全没有毒害作用的特点,已成为目前世界上应用最广泛地微生物杀虫剂。自从 1987 年比利时的Vaeck等[18]报道了首例转Bt基因烟草的研究结果以来,Bt毒素蛋白基因已在烟草、棉花(Gossypiumspp.)、水稻(Oryzasativa)、玉米(Zeamays)、番茄(Solanumlycopersicum)、甘蓝(Brassicaoleracea)等50余种植物上获得成功[19-20]。Cooper等[21]已经成功获得抗马铃薯甲虫的转Bt基因马铃薯,国内也有少数相关的研究报道[22]。尽管运用现代基因工程技术得到了大量的转Bt基因植物,然而随着转Bt毒蛋白基因植物的大面积种植,给昆虫造成很高的选择压力,可能会使昆虫对此毒蛋白产生抗性,这些不容忽视的问题限制了这些转基因植物向商品化、实用化方向发展。为此,本研究运用CaMV 35S强组成型启动子驱动CryⅢA基因来提高Bt毒蛋白基因的表达量来提高转基因植株的抗虫性;同时,本研究充分考虑到基因表达的时空特异性,选用马铃薯茎叶特异表达启动子ST-LS1,使目的基因CryⅢA基因只限定在容易被马铃薯甲虫危害的叶片和茎秆中特异表达,从而实现对目的基因的表达调控,而目的基因在块茎中不表达,从而使人们食用转基因马铃薯更加安全。本研究通过qRT-PCR检测分析表明在CaMV 35S和ST-LS1启动子驱动的CryⅢA转基因马铃薯植株中,CryⅢA基因在叶中的表达量最高,且在ST-LS1启动子驱动的CryⅢA转基因植株的根中未见表达。对于转基因植株抗虫效果的鉴定正在进行中。
4 结论
采用In-Fusion技术成功构建了组成型启动子CaMV 35S和马铃薯茎叶特异表达启动子ST-LS1驱动的CryⅢA基因植物表达载体,转化马铃薯栽培品种获得了转基因植株,应用组织特异性表达启动子ST-LS1实现了目的基因的定向表达,为丰富转基因理论和培育抗甲虫转基因马铃薯新品系奠定了基础。
猜你喜欢
学与玩(2022年10期)2022-11-23
今日农业(2022年3期)2022-06-05
少儿科学周刊·儿童版(2021年21期)2021-12-11
今日农业(2021年11期)2021-11-27
今日农业(2021年4期)2021-06-09
江西农业学报(2021年4期)2021-04-20
陕西画报(2016年1期)2016-12-01
创新科技(2015年1期)2015-12-24
西南医科大学学报(2015年1期)2015-08-22
中国当代医药(2015年9期)2015-03-01
