
11β-羟化酶缺陷症遗传学研究现状
2014-11-26 08:23王晓晶聂敏孙梅励
生殖医学杂志 2014年2期
王晓晶,聂敏,孙梅励
(中国医学科学院,北京协和医学院,北京协和医院内分泌科;卫生部内分泌重点实验室,北京 100730)
先天性肾上腺皮质增生症(congenital adrenal hyperplasia,CAH)是一组由皮质醇生物合成过程中某一种酶缺陷引起的常染色体隐性遗传病。11β-羟化酶缺陷症(11β-Hydroxylase deficiency,11β-OHD)是仅次于21-羟化酶缺陷症(21α-OHD)引起CAH的第二大病因,约占CAH的5%~8%。其在普通人群中的发病率为1/100,000~1/200,000活产儿,而在近亲结婚较普遍的从摩洛哥移民到以色列的犹太人群中,发病率高达1/7,000~1/5,000活产儿[1]。
11β-OHD 是由 CYP11B1基因突变所致,CYP11B1基因突变使P450c11B1的11β-羟化酶活性受损,11-去氧皮质醇转化为皮质醇受阻,合成皮质醇的前体物质:11-去氧皮质酮(DOC)、17-羟孕酮等大量堆积,同时转而合成过多的雄激素。血清高水平的DOC由于其具有弱盐皮质激素的作用,导致水钠潴留,血容量增加,抑制肾素的合成,造成约2/3的经典型11β-OHD患者有低肾素性高血压[2];而过多的雄激素引起男性患儿性早熟以及女性外生殖器男性化。然而,非经典型11β-OHD患者,血压一般正常或仅有轻度升高,女性患者出生时外生殖器正常,青春期前后可出现阴蒂肥大等雄激素过多症状,成年妇女可有多毛、月经稀发等表现。
近年来,随着分子生物学技术的发展以及对新生儿优生优育的重视,目前有关CYP11B1基因新突变位点的报道增多并随之对其功能的研究亦逐渐增加。本文将综述11β-羟化酶缺陷症分子遗传学的研究现状。
一、11β-OHD分子遗传基础
11β-OHD为常染色体隐性遗传病,其致病基因为CYP11B1。CYP11B1定位于8号染色体长臂2区1带(8q12),总长度为6.03kb,共含有9个外显子,编码由503个氨基酸组成的P450c11B1蛋白;CYP11B2编码醛固酮合成酶,位于CYP11B1上游约45kb处,与CYP11B1具有高度同源性,二者的外显子和内含子的相似度分别高达95%和90%,两者的主要区别在于CYP11B2基因第5内含子多插入442个碱基对[3]。CYP11B1基因突变,使11β-羟化酶活性减弱或丧失,导致11β-羟化酶缺陷症。White等[4]于1991年首次报道了CYP11B1基因突变(R448H)引起11β-OHD。到目前为止,文献报道的CYP11B1基因突变已达70余种,包括错义突变、无义突变、插入、缺失以及剪切位点的突变等,这些突变位点在所有外显子区均有分布,但主要集中在第2、3、6、7、8外显子区[3](图1),推测与这些外显子区富含GpC位点以及其所对应的氨基酸相对保守有关。我室对12例就诊于北京协和医院,初步诊断为11β羟化酶缺陷症的患者行CYP11B1基因突变检测,发现了9种CYP11B1基因新突变,包括4种错义突变,2种无义突变以及剪切位点突变,纯合插入突变和缺失突变各一种,且主要集中在第3和第8外显子区(文章待发表),与已报道的研究[3]结果一致。

图1 已知CYP11B1基因突变类型简图
二、CYP11B1基因突变对P450c11B1酶活性的影响
1.CYP11B1基因错义突变与酶活性:错义突变是CYP11B1基因突变最常见的类型,目前已报道50余种,主要通过蛋白三维结构模拟和体外蛋白表达实验研究其对酶活性的影响。P450c11B1酶作为P450c11的一种,以血红素为辅基催化氧化还原反应,其三维结构模拟图(图2)显示I~L螺旋(helix)属于高度保守的血红素结合区,其中第450位的半胱氨酸(C450)巯基与血红素铁原子相结合,形成酶活性位点之一[3,5],因此,C450邻近的氨基酸改变,均会影响血红素与酶的结合,使酶活性丧失。体外研究也证实,R427H、V441G、G444D、G446V、R448H、R453Q、R454C突变均使酶活性丧失[6-9],其中R454C突变仅在中国人群中发现。此外,I螺旋是底物“结合袋”的一部分,含有许多疏水性氨基酸以及潜在的酶活性位点,参与底物的识别与结合,研究显示I螺旋构像轻微的改变,即可导致P450c11B1酶活性严重受损[10]。脯氨酸取代亮氨酸(L299P)改变了I螺旋的空间位置,血红素极性与非极性部分的空间位置也发生重排,使酶活性降至(1.6±0.8)%[11]。368位的丙氨酸(A368)与V336、L340相互作用维持其周围的疏水环境,酸性天冬氨酸取代中性丙氨酸(A368D)同样会改变I螺旋的构像,使酶活性降至(1.17±1.9)%[10]。同理,P94L改变K-L环的方向[10];位于K螺旋、I螺旋的A331V、E371G突变改变了酶的3级结构[12],均使酶活性丧失。
F和G螺旋以及G螺旋和I螺旋间的B-C环是底物进入酶活性中心与其结合的潜在通道,尤其G螺旋是底物初始识别所必需的。A259位于G螺旋,A259D突变打破了丙氨酸周围的疏水环境,使酶与底物的结合障碍,酶活性受损[6]。W116位于B-C环,116位色氨酸被取代,同样使酶与底物结合受阻,酶活性丧失,目前发现该位点有 W116X、W116G、W116C三种突变[13],高度保守的 T318同样已发现 T318R、T318M、T318P三种突变[12],表明W116和T318位点可能不稳定,易于发生突变。此外,V129M、V148G、R384Q、R384G等突变均因影响了酶与底物的结合,使酶活性下降。
总之,CYP11B1基因错义突变均不同程度的影响了P450c11B1酶的空间构象,尤其是维持酶活性关键区的构象,从而影响了酶活性。对发现的突变位点进行蛋白三维结构的模拟将有助于合理解释和进一步证实体外蛋白表达实验的研究结果。
2.CYP11B1基因无义突变和框移突变与酶活性:目前已报道10余种CYP11B1基因无义突变。无义突变使终止密码子提前出现,蛋白质翻译提前终止,导致截短蛋白产生,若维持酶活性的重要氨基酸(如血红素结合区的第443位至463位之间氨基酸)缺失,将使酶活性丧失。已发现的CYP11B1基因无义突变均使血红素结合区的氨基酸缺失。
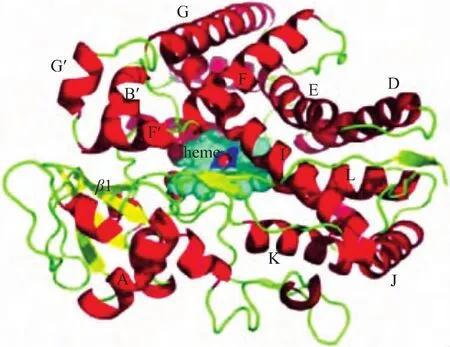
图2 P450c晶体结构[5]
插入或缺失突变使阅读框架移动,相应的氨基酸序列改变,翻译出错误蛋白,同时提前出现终止密码子,蛋白表达提前终止,使酶蛋白的相应功能结构域发生改变或缺失,酶活性丧失。如最新报道的框移突变g.2697del449、c.652-653insT、g.4671-4672insG、c.1359-1360insG分别使第165位的丙氨酸、218位的丝氨酸、404位精氨酸和第454位精氨酸分别转变为丙氨酸、苯丙氨酸、脯氨酸和丙氨酸,并且蛋白表达分别于第217、258、421和468位提前终止[1,14-16],高度保守的血红素结合区氨基酸缺失或改变,酶活性丧失。
3.CYP11B1基因剪切位点突变与酶活性:关于CYP11B1基因剪切位点突变的报道较少。剪切位点突变会导致mRNA成熟前的错误剪切,可以表现为原有的剪切信号(5’GT-AG3’)隐藏、潜在的剪切位点激活形成新的剪切位点或直接跳过整个外显子在下一个外显子剪切信号处剪切等,从而形成错误的转录本,表达出错误蛋白,使酶活性丧失。如IVS8+4A>G突变,剪切时跳过第8外显子,使成熟的mRNA缺乏第8外显子,酶活性丧失。同理,IVS7+4A>G、IVS7-9C>A、IVS5+2T>G等剪切位点的突变[5,15,17],均因错误剪切而使酶活性丧失。
4.CYP11B1基因嵌合突变与酶活性:由于CYP11B1与CYP11B2基因高度同源,减数分裂时,两者可发生不平等等位基因交换,产生CYP11B2-CYP11B1嵌合基因,其启动子区由CYP11B2提供,受血管紧张素II(AngⅡ)和K+的调节,尽管体外研究显示,该嵌合基因编码的蛋白质具有11β-羟化酶的活性,但由于CYP11B2只在肾上腺球状带表达,因此,不能催化束状带11-去氧皮质醇转化为皮质醇。若患者为CYP11B2-CYP11B1嵌合基因纯合子或为含有CYP11B2-CYP11B1嵌合基因的复合杂合子,则表现为11β-羟化酶缺陷症。目前,共报道了3例由CYP11B2-CYP11B1嵌合基因引起的11β-OHD。Hampf 等[18]首次报道了一例携带CYP11B2-CYP11B1嵌合基因的复合杂合子患者,CYP11B2-CYP11B1嵌合基因由CYP11B2的1~4外显子和CYP11B1的5~9外显子组成,该患者另一条等位基因携带IVS3+16G>T剪切位点突变;Kuribayashi 等[19]报道一例高加索患者为CYP11B2-CYP11B1/G314R复合杂合子,嵌合基因由CYP11B2的1~3外显子和CYP11B1的4~9外显子组成;另一例为CYP11B2-CYP11B1纯合突变患者,该嵌合基因由CYP11B2的1~6外显子和CYP11B1的7~9外显子组成[20]。对临床表现符合11β-羟化酶缺陷症的患者,如果用正常引物无法扩增出CYP11B1基因,应想到CYP11B2-CYP11B1纯合突变的可能性。
5.基因型与表型关系:非经典型11β-OHD患者可无或有轻度高血压,或有轻度的雄激素过多的表现,研究发现这些患者不典型的临床表现与其CYP11B1基因突变对酶活性的影响相对较小有关。Joehrer等[21]首次对两例非经典型11β-OHD 患者进行了分子遗传学研究,该两例患者分别为N133H/T319M和P42S/Y423X复合杂合子,体外实验发现P42S、N133H、T319M突变使酶活性保留15%~40%。此后,Peters等[22]报道了一例L489S纯合突变的非经典型患者,蛋白质三维结构模拟结果示L489S仅轻度改变了酶与底物的亲和力。2010年,Parajes等[13]在非经典型患者中检测到的M88I、P159L突变分别使酶活性下降至野生型的40%和25%。而研究显示,引起经典型11β-OHD的CYP11B1突变通常使酶活性下降至5%以下或使酶活性完全丧失,但对于每个特定突变,其与患者临床表现的严重程度无明显相关性,携带相同基因突变的不同患者,可有轻度或重度高血压,雄激素过多的表现也轻重度不等。因此,基因型与表型之间的相关性,尚有待进一步研究。
综上所述,11β-OHD为CAH第二常见类型,它是由CYP11B1基因突变引起的常染色体隐性遗传病,男性化和高血压为经典型患者主要的临床表现。目前,有关CYP11B1基因突变位点的报道已达70余种,基因突变对酶活性影响的研究也逐步深入。尽管研究发现非经典型患者的CYP11B1基因突变对酶活性的影响相对较小,但对于某个特定CYP11B1基因突变,其与患者临床表现的严重程度无明显相关性。对CYP11B1基因突变及其功能的深入研究,将有助于进一步揭示基因型与表型的关系,同时也为产前诊断及遗传咨询提供重要的依据,以提高婴儿的优生优育水平。
[1]Ben C I,Riepe F G,Kahloul N,et al.Two novel CYP11B1 mutations in congenital adrenal hyperplasia due to steroid 11beta hydroxylase deficiency in a Tunisian family[J].Gen Comp Endocrinol,2012,175:514-518.
[2]Kharrat M,Trabelsi S,Chaabouni M,et al.Only two mutations detected in 15Tunisian patients with 11betahydroxylase deficiency:the p.Q356Xand the novel p.G379V[J].Clin Genet,2010,78:398-401.
[3]Nguyen HH,Nguyen TH,Vu CD,et al.Novel homozygous p.Y395Xmutation in the CYP11B1gene found in a Vietnamese patient with 11beta-hydroxylase deficiency[J].Gene,2012,509:295-297.
[4]White PC,Dupont J,New MI,et al.A mutation in CYP11B1(Arg-448-His)associated with steroid 11beta-hydroxylase deficiency in Jews of Moroccan origin[J].J Clin Invest,1991,87:1664-1667.
[5]Anzenbacher P,Anzenbacherová E,Lange R,et al.Active sites of cytochromes P450:What are they like?[J].Acta Chim Slov,2008,55:63-66.
[6]Chabraoui L,Abid F,Menassa R,et al.Three novel CYP11B1 mutations in congenital adrenal hyperplasia due to steroid 11Beta-hydroxylase deficiency in a moroccan population[J].Horm Res Paediatr,2010,74:182-189.
[7]Wu C,Zhou Q,Wan L,et al.Novel homozygous p.R454C mutation in the CYP11B1gene leads to 11beta-hydroxylase deficiency in a Chinese patient[J].Fertil Steril,2011,95:1122-1123.
[8]Krone N,Grotzinger J,Holterhus PM,et al.Congenital adrenal hyperplasia due to 11-hydroxylase deficiency-insights from two novel CYP11B1mutations(p.M92X,p.R453Q)[J].Horm Res,2009,72:281-286.
[9]Ye ZQ,Zhang MN,Zhang HJ,et al.A novel missense mutation,GGC(Arg454)>TGC(Cys),of CYP11B1gene identified in a Chinese family with steroid 11beta-hydroxylase deficiency[J].Chin Med J(Engl),2010,123:1264-1268.
[10]Krone N,Grischuk Y,Muller M,et al.Analyzing the functional and structural consequences of two point mutations(P94Land A368D)in the CYP11B1gene causing congenital adrenal hyperplasia resulting from 11-hydroxylase deficiency[J].J Clin Endocrinol Metab,2006,91:2682-2688.
[11]Riedl S,Nguyen HH,Clausmeyer S,et al.A homozygous L299Pmutation in the CYP11B1gene leads to complete virilization in 46,XX individuals with 11-beta-hydroxylase deficiency[J].Horm Res,2008,70:145-149.
[12]Zhao LQ,Han S,Tian HM.Progress in molecular-genetic studies on congenital adrenal hyperplasia due to 11betahydroxylase deficiency[J].World J Pediatr,2008,4:85-90.
[13]Parajes S,Loidi L,Reisch N,et al.Functional consequences of seven novel mutations in the CYP11B1gene:four mutations associated with nonclassic and three mutations causing classic 11{beta}-hydroxylase deficiency[J].J Clin Endocrinol Metab,2010,95:779-788.
[14]Xu C,Qiao J,Liu W,et al.Identification and functional characterization of a large deletion of the CYP11B1gene causing an 11beta-Hydroxylase deficiency in a Chinese pedigree[J].Horm Res Paediatr,2012,78:212-217.
[15]Zhang M,Liu Y,Sun S,et al.A prevalent and three novel mutations in CYP11B1gene identified in Chinese patients with 11-beta hydroxylase deficiency[J].J Steroid Biochem Mol Biol,2013,133:25-29.
[16]Soardi FC,Penachioni JY,Justo GZ,et al.Novel mutations in CYP11B1gene leading to 11beta-hydroxylase deficiency in Brazilian patients[J].J Clin Endocrinol Metab,2009,94:3481-3485.
[17]Dumic K,Wilson R,Thanasawat P,et al.Steroid 11-beta hydroxylase deficiency caused by compound heterozygosity for a novel mutation in intron 7(IVS 7DS+4Ato G)in one CYP11B1allele and R448Hin exon 8in the other[J].Eur J Pediatr,2010,169:891-894.
[18]Hampf M,Dao NT,Hoan NT,et al.Unequal crossing-over between aldosterone synthase and 11beta-hydroxylase genes causes congenital adrenal hyperplasia[J].J Clin Endocrinol Metab,2001,86:4445-4452.
[19]Kuribayashi I,Nomoto S,Massa G,et al.Steroid 11-betahydroxylase deficiency caused by compound heterozygosity for a novel mutation,p.G314R,in one CYP11B1allele,and a chimeric CYP11B2/CYP11B1in the other allele[J].Horm Res,2005,63:284-293.
[20]Nimkarn S,New MI.Steroid 11beta-hydroxylase deficiency congenital adrenal hyperplasia[J].Trends Endocrinol Metab,2008,19:96-99.
[21]Joehrer K,Geley S,Strasser-Wozak EM,et al.CYP11B1 mutations causing non-classic adrenal hyperplasia due to 11 beta-hydroxylase deficiency[J].Hum Mol Genet,1997,6:1829-1834.
[22]Peters CJ,Nugent T,Perry LA,et al.Cosegregation of a novel homozygous CYP11B1mutation with the phenotype of nonclassical congenital adrenal hyperplasia in a consanguineous family[J].Horm Res,2007,67:189-193.
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
中华实用诊断与治疗杂志(2022年2期)2022-09-02
中国生殖健康(2020年4期)2021-01-18
中国生殖健康(2020年2期)2021-01-18
热带作物学报(2020年9期)2020-10-29
中国现代医生(2020年12期)2020-07-04
中国生殖健康(2018年4期)2018-11-06
小学生导刊(2018年13期)2018-06-29
中国生殖健康(2018年2期)2018-01-12
江苏农业科学(2015年10期)2015-12-23
