
四种药用异黄酮的全合成
2015-01-08 08:10乔金凤张尊听
天然产物研究与开发 2015年6期
魏 涛,乔金凤,王 丁,韦 威,贺 云,张尊听
教育部药用资源与天然药物化学重点实验室西北濒危药材资源开发国家工程实验室 陕西师范大学化学化工学院,西安 710062
异黄酮类化合物主要存在于大豆、红三叶草、野葛等豆科植物中。现代药理研究表明,该类化合物具有抗肿瘤[1]、抗病毒[2]、抗菌[3]、消炎[4]、治疗心脑血管疾病[5]等药理作用和生物活性,是许多中草药的有效成分之一。目前获得异黄酮的方法主要有天然植物提取法和化学合成法。由于提取法存在溶剂消耗量大、溶剂残留、能耗大、产率低以及难以解决重金属残留等缺点,因此进行异黄酮类化合物的合成研究具有重要的意义。文献报道,合成异黄酮主要方法是苯基苄基酮途径[6](又叫脱氧安息香途径)和重排查尔酮途径[7],此外还有偶联反应[8-10]和邻羟基苯甲醛和烯胺的缩合反应[11]等方法。苯基苄基酮途径包括两大步反应:一是制备中间体脱氧安息香,另一步是脱氧安息香的环合反应。根据所用原料、试剂及反应过程,有四种合成脱氧安息香的方法,它们分别是苯乙腈法[12]、苯乙酰氯法[13]及Fries 重排法(图1)[14]。脱氧安息香的增碳关环反应方法研究较多,可以用脱氧安息香分别与烷基草酰卤[15]、DMF/甲基磺酰氯[16]、DMF/五氯化磷[17]、N-甲醛基咪唑反应构成异黄酮环[18]。但是采用苯基苄基酮途径合成异黄酮,所用反应条件剧烈、试剂毒性较大、反应时间长、产物纯化比较复杂、产率较低。

图1 脱氧安息香途径合成异黄酮Fig.1 Synthesis of isoflavones by the deoxybenzoin route
20 世纪70 年代,出现了查尔酮途径合成异黄酮,主要是以取代苯乙酮和取代苯甲醛为原料,经Claisen-Schemidt 缩合生成相应的查尔酮,查尔酮在重金属铊盐作用下经过重排反应合成异黄酮(图2)[19],该方法产率较低且生产工艺中大量使用铊盐;还可以将查尔酮利用环氧化重排法制备异黄酮[20],但是产率较低。此外,黄烷酮氧化重排制备异黄酮也是制备异黄酮的方法之一[21],多以铊盐和高碘化物作催化剂,但催化剂和溶剂对反应产率影响较大。

图2 金属铊的作用下经查尔酮途径合成异黄酮Fig.2 Synthesis of isoflavones by the chalcone route with Thallium
随着偶联反应技术的出现,Yoke[8]、Vasselin[9]和Rao[10]分别报道了钯催化的Suzuki 偶联反应、Stille 有机锡偶联反应和Stille 有机铋偶联反应合成异黄酮化合物的新方法(图3)。近年来,我们曾完成了钯/碳催化下3-碘色原酮与四芳基锡偶联反应合成异黄酮[22]。偶联反应合成异黄酮,都是以容易制备的3-碘色原酮为原料,反应条件温和,原子利用率高于苯基苄基酮和查尔酮途径,产率可达80%~90%。
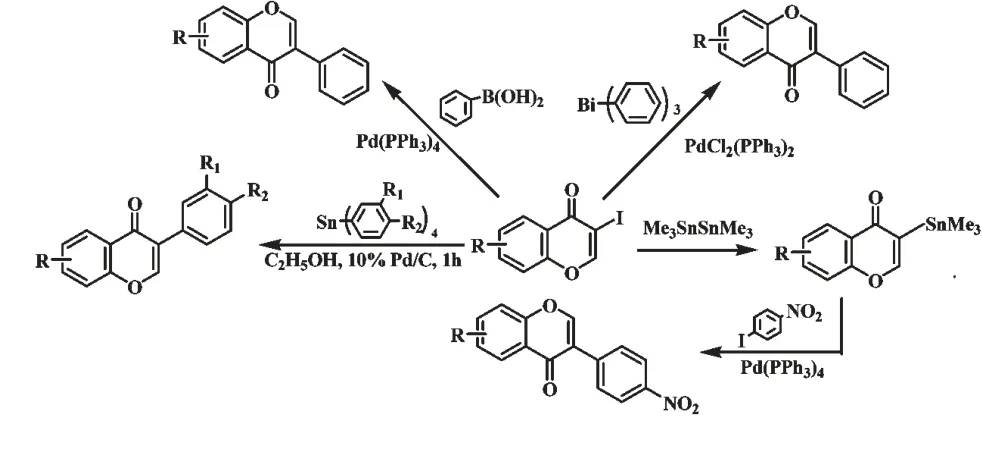
图3 钯催化偶联反应合成异黄酮Fig.3 Synthesis of isoflavones by Pd-catalyzed cross-coupling reactions
Leo 和Heinz[11]研究了邻羟基苯甲醛和烯胺的缩合反应,用取代邻羟基苯甲醛与N-苯乙烯基吗琳反应合成异黄酮化合物。该方法合成异黄酮和8-甲氧基异黄酮的产率分别为46.2%和42.8%,反应要求无水溶剂和氮气保护,而且成本较高。Thomsen等还报道了以水杨醛肟与烯胺(N-苯乙烯基玛琳)反应合成异黄酮[23],需经乙酰化、还原、环合反应得到异黄酮化合物,产率为37%,反应过程复杂,产率低且需用薄层色谱法进行分离纯化,因此工业应用价值较低。本文在我们研究Negishi 交叉偶联反应合成异黄酮的基础上[24],分别以2,4-二羟基苯乙酮(1a)、2,4,6-三羟基苯乙酮(1c)和对溴苯酚(8)、对甲氧基溴苯(9a)为起始原料,利用Negishi 交叉偶联反应合成芒柄花素(7a)、大豆苷元(7b)和鹰嘴豆芽素A(7c)、染料木素(7d)四种药用异黄酮化合物,但由于锌试剂(5)无法容忍活泼氢(如羟基)的存在,因此首先需要对1a 4 位的羟基、1c 4,6 位的羟基和8 的羟基用MOMCl(氯甲基甲醚)进行保护;再将2 与DMF-DMA(N,N-二甲基甲酰胺二甲缩醛)缩合及与碘关环合成4a、4c;最后再通过4 和5 的Negishi 交叉偶联和水解反应合成7a~7d(图4)。
1 材料与方法
1.1 试剂与仪器
2,4-二羟基苯乙酮(1a),2,4,6-三羟基苯乙酮(1c)(北京恒业中远化工有限公司,纯度>97%);对溴苯酚(8)、对甲氧基溴苯(9a),(上海柏卡化学技术有限公司,纯度>98%);DMF-DMA(N,N-二甲基甲酰胺二甲缩醛)(百灵威科技有限公司,纯度>98%);锌粉,溴化钴,氯丙烯,三氟乙酸,溴代芳烃(阿拉丁试剂,纯度>98%);NiCl2(PPh3)2,PPh3(上海柏卡化学技术有限公司);乙腈,四氢呋喃,DMF(N,N-二甲基甲酰胺),石油醚,乙酸乙酯,甲醇、乙醇,氯仿等常用试剂均为分析纯。
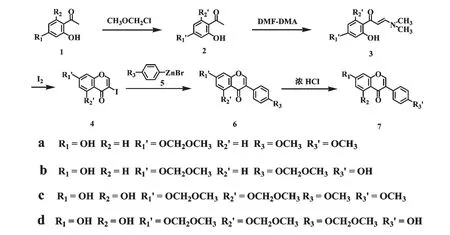
图4 7a-7d 的全合成路线Fig.4 Total synthetic strategy of 7a-7d
X-5 显微熔点测定仪(控温型,温度未经校正);ZF-2 型三用紫外仪;Bruker 170SX FT-IR 红外光谱仪(KBr 压片法);Bruker AM-300 超导傅立叶数字化核磁共振仪(TMS 为内标);Bruker MAXIS 型高分辨质谱仪。
TLC 薄层层析硅胶GF254(400 目)和柱层析硅胶(200~300 目)均为为青岛海浪化工厂生产;TLC薄层层析硅胶检测用254 nm 和365 nm 紫外灯。
1.2 合成部分
1.2.1 芳基锌试剂的制备
对甲氧基甲醚基溴苯(9b)的制备:称取8(47.1 mmol,8.1 g)于500 mL 圆底烧瓶中,并向其中加入200 mL 丙酮为溶剂,在常温搅拌条件下加入无水碳酸钾(84.8 mmol,11.7 g),58 ℃水浴回流15 min后,快速称取MOMCl(51.8 mmol,4.2 g)于盛有47 mL 丙酮溶剂(稀释MOMCl)的100 mL 圆底烧瓶中,然后将其倒入连接于反应瓶上方的恒压滴液漏斗中,缓慢逐滴滴加至完毕,回流反应1.5 h,TLC 检测原料8 几乎消耗完全,停止反应。反应物减压蒸馏回收溶剂,加入适量水,用二氯甲烷萃取,分出有机相并用无水MgSO4干燥,减压浓缩得到9b 粗品(40.0 mmol,8.7 g),产率为85%。
芳基锌试剂5 的制备[25]:称取锌粉(60 mmol,3.9 g)和溴化钴(2.1 mmol,0.7 g)于50 mL 圆底烧瓶中,并向其中加入16 mL 乙腈,在搅拌使其混匀的同时依次加入氯丙烯(6.3 mmol,0.48 mL)和三氟乙酸(0.40 mL),发现反应液颜色由原来的蓝绿色逐渐加深变为深棕色甚至黑色,同时会有气体生成且反应放热。常温搅拌5 min 后,加入40.0 mmol溴代芳烃(9a,9b)常温搅拌1 h,停止反应(图5),得芳基锌试剂5,产率80%~90%。依据文献[26]对芳基锌试剂的浓度进行了测定。
1.2.2 7a 和7b 的合成
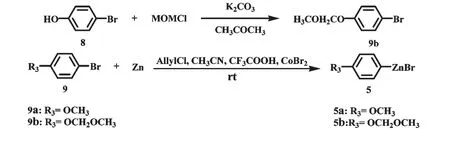
图5 芳基锌试剂5a 和5b 的合成Fig.5 Synthesis of arylzinc reagents 5a and 5b
1.2.2.1 称取1a(26.4 mmol,4.0 g)于500 mL 圆底烧瓶中,并向其中加入120 mL 丙酮为溶剂,在常温搅拌条件下,加入无水碳酸钾(47.5 mmol,6.6 g),58 ℃水浴回流15 min 后,快速称取MOMCl(29.0 mmol,2.3 g)于盛有20 mL 丙酮溶剂(稀释MOMCl)的50 mL 圆底烧瓶中,然后将其倒入连接于反应瓶上方的恒压滴液漏斗中,缓慢逐滴滴加至完毕,回流反应1.5 h,TLC 检测原料1a 几乎消耗完全,停止反应。将反应液减压蒸馏,回收溶剂,加入适量水用二氯甲烷萃取,分出有机相并用无水MgSO4干燥,减压浓缩得到2a 粗品4.7 g,产率为91%。
将2a(24.0 mmol,4.7 g)粗品盛于500 mL 圆底烧瓶中,并向其中加入DMF-DMA(N,N 二甲基甲酰胺二甲缩醛)(48.0 mmol,5.7 g)和200 mL DMF 为溶剂,80 ℃水浴加热搅拌1 h,TLC 检测至原料反应完全,停止反应。待反应液冷却至室温,将其倒入400 mL 饱和食盐水中,静置片刻,发现有金黄色针状晶体析出,抽滤,用蒸馏水将滤饼洗至中性,烘干得3a 粗品5.3 g,产率88%;按照文献[24]的方法,将3a 粗品(21.1 mmol,5.3 g)与碘(31.5 mmol,10.6 g)在260mL 氯仿溶剂中室温搅拌2 h,TLC 检测原料消耗完全,停止反应。向反应液中加入100 mL 5%亚硫酸钠溶液,搅拌至反应液由深红色变为淡黄色甚至褪至无色之后,反应猝灭完全,分离出有机相,将水相用氯仿萃取三次,合并有机相,用蒸馏水洗有机相至中性,无水MgSO4干燥,抽滤,减压蒸干溶剂即得4a,5.7 g,产率82%。
1.2.2.2 称取4a(17.3 mmol,5.7 g)、4 ×10-3mol%NiCl2(PPh3)2和无水LiCl(26.0 mmol,1.1 g)于500 mL 圆底烧瓶中,并向其中加入86 mL THF 溶剂,室温搅拌5 min 后,加入34.6 mmol 新制备的芳基锌试剂(5a),室温搅拌1 h,TLC 检测原料几乎消耗完全,加2 mL 水停止反应。先将反应液中的溶剂减压蒸干,加入86 mL 10%盐酸,200 mL MeOH 溶剂,68℃回流30 min,TLC 检测原料消耗完全,停止反应。将反应液用400 mL 氯仿萃取两次,合并有机相,用蒸馏水洗至中性,无水MgSO4干燥,过滤,减压蒸馏除去溶剂,经乙醇重结晶,得到芒柄花素(7a)纯品4.2 g,产率90%。按照合成7a 的方法,以5b 代替5a 合成大豆苷元(7b),得到纯品3.6 g,产率为81%。
化合物7a 白色晶体;mp.257~258 ℃;IR(KBr)νmax3697,3078,2985,2835,1638,1600,1513,1453,1386,1316,1273,1249,1177,1100,1025,887,840,813,780 cm-1;1H NMR (DMSO,300 MHz)δ 10.74 (1H,s),8.24 (1H,s),7.90 (1H,d,J=8.7 Hz),7.43 (2H,d,J=8.5 Hz),6.86 (4H,m),3.71(3H,s);13C NMR (DMSO,75 MHz)δ:174.6,162.5,158.9,157.4,153.01,30.0,127.3,124.2,123.1,116.6,115.1,113.6,102.1,55.1;HR-EI-MS m/z[M+Na]+(calcd for C16H12O4Na:291.0633,found:291.0649)。
化合物7b 白色晶体;mp.304~305 ℃;IR(KBr)νmax3509,1941,1816,1710,1616,1536,1499,1417,1323,1296,1267,1207,1112,1059,1029,971,913,862,805,762,704,564,536,515 cm-1;1H NMR (CDCl3,400 MHz)δ 10.70 (1H,s),9.45(1H,s),8.19 (1H,m),7.89 (1H,d,J=8.8 Hz),7.39-7.24 (2H,m),6.85 (1H,dd,J=8.8,2.2Hz),6.78 (1H,d,J=2.0 Hz),6.74 (2H,d,J=8.6 Hz,);13C NMR (CDCl3,100 MHz)δ 179.9,167.7,162.6,162.4,158.0,157.9,135.3,132.5,128.7,127.8,121.9,120.3,120.2,107.3;HR-EIMS m/z [M+Na]+(calcd for C15H10O4Na:277.0477,found:277.0499)。
1.2.3 7c 和7d 的合成
1.2.3.1 称取1c(23.8 mmol,4.0 g)于500 mL 圆底烧瓶中,并向其中加入240 mL 丙酮为溶剂,在常温搅拌条件下,加入无水碳酸钾(85.6 mmol,11.8 g),58 ℃水浴回流15 min 后,快速称取MOMCl(52.4 mmol,4.2 g)于已装有50 mL 丙酮溶剂(稀释MOMCl)的100 mL 圆底烧瓶中,然后将其倒入连接于反应瓶上方的恒压滴液漏斗中,缓慢逐滴滴加至完毕,回流反应1.5 h,TLC 检测原料几乎消耗完全,停止反应,按照1a 的后处理方法进行处理,即得2c粗品,5.3 g,产率87%。同于3a 制备方法,合成3c粗品,5.7 g,产率89%;类似于4a 制备方法,将其中氯仿溶剂换为甲醇[27],室温下反应2 h 即可得到4c,5.9 g,产率81%。
1.2.3.2 称取4c(14.9 mmol,5.9 g)、4 ×10-3mol%NiCl2(PPh3)2和无水LiCl(22.4 mmol,0.9 g)于500 mL 圆底烧瓶中,并向其中加入75 mL THF 溶液,室温搅拌5 min 后,加入33 mmol 新制备的芳基锌试剂(5a)室温搅拌1 h,TLC 检测原料几乎消耗完全,加2 mL 水停止反应。先将反应液中的溶剂减压蒸干,再向反应液中加入70 mL 浓盐酸,180 mL MeOH溶剂,68 ℃回流1 h,TLC 检测原料消耗完全,停止反应。反应液用370 mL 氯仿萃取两次,合并有机相,用蒸馏水洗至中性,无水MgSO4干燥,过滤,减压蒸馏除去溶剂,经乙醇重结晶得到鹰嘴豆芽素A(7c)纯品,3.7 g,产率为88%。按照合成7c 的方法,以5b 代替5a,合成染料木素(7d),得到纯品2.0 g,产率为49%。
化合物7c 白色晶体;mp.211~213 ℃;IR(KBr)νmax3515,3355,3111,1723,1636,1592,1537,1482,1379,1321,1279,1210,1156,1074,1037,941,894,862,815,786,753,713,679,578,547,525,483 cm-1;1H NMR(DMSO,300 MHz)δ 12.91 (1H,s),10.87 (1H,s),8.35 (1H,s),7.49(2H,d,J=8.4 Hz),6.99 (d,J=8.3 Hz,2H),6.38(s,1H),6.22 (s,1H),3.78 (s,3H);13C NMR(DMSO,100 MHz)δ 180.1,164.3,162.0,159.1,157.6,154.2,130.1,122.9,121.9,113.7,104.4,99.0,93.7,55.1;HR-EI-MS m/z[M +Na]+(calcd for C16H12O5Na:307.0582,found:307.0606)。
化合物7d 白色晶体;mp.299~300 ℃;IR(KBr)νmax3410,3105,1816,1652,1614,1568,1503,1427,1394,1360,1312,1256,1204,1176,1143,1063,1042,912,883,843,816,788,744,641,613,568,532,488,439 cm-1;1H NMR (DMSO,300 MHz)δ 12.86 (1H,s),10.79 (1H,s),9.51 (1H,s),8.20 (1H,s),7.28 (2H,d,J=8.4 Hz),6.73(2H,d,J=8.4 Hz),6.20 (2H,m);13C NMR (DMSO,75 MHz)δ 180.2,164.2,162.0,157.5,157.4,153.8,130.1,122.3,121.2,115.0,104.4,98.9,93.6;HR-EI-MS m/z [M+Na]+(calcd for C15H10O5Na:293.0426,found:291.0450)。
2 结果与讨论
2.1 碱种的种类及用量、溶剂、温度、原料配比对活泼羟基保护反应的影响
以1a 合成1b 为例,依据文献[28],以DMF 为溶剂,NaH 为碱,在冰浴(0 ℃)中进行反应,产率可达82%(表1,Entry 1);但由于NaH 容易吸水,易爆炸,较危险,不利于实验的扩大化,且溶剂DMF 沸点较高,不利于反应的后处理。因此选用实验室常用的K2CO3为碱,分别以DMF、乙醇、丙酮为溶剂进行反应,实验结果表明,以乙醇为溶剂,该反应几乎不能进行(表1,Entry 3);以DMF 和丙酮为溶剂反应均可以进行且产率分别为69%和74%(表1,Entries 2,4)。故以丙酮为溶剂,K2CO3为碱。再对各个反应物的摩尔比进行优化,实验结果表明:1a∶MOMCl∶K2CO3摩尔比为1 ∶1.1 ∶1.8 时,产物的产率与1∶1.1∶2.0 或1∶1.2∶1.8 时的产率相差不大,再继续增大K2CO3的量或者MOMCl 的量对产物的产率影响也不大(表3,Entries 5-8),而若继续减小K2CO3的量或者MOMCl 的量,产物产率会明显降低(表1,Entries 9,10)。通过实验,确定该反应的最佳条件是:以K2CO3为碱,1a∶MOMCl∶K2CO3摩尔比为1∶1.1 ∶1.8,在丙酮溶剂中回流2 h,产率可达90%。
2.2 催化剂种类、配体及其用量对Negishi 偶联反应的影响
以4a 和5a 的偶联反应为例优化反应条件。依据文献[24],选用NiCl2(PPh3)2为催化剂,用量为5×10-3mol%,不加入配体,对4a 的量进行筛选,发现其与5a 的摩尔比从1∶1.2 逐渐增至1∶2 时,产物产率逐渐增至92%(表2,Entries 1-3),故将4a 与5a的摩尔比确定为1∶2;随后将催化剂的用量从5 ×10-3mol%逐渐降低,发现当催化剂用量降为4 ×10-3mol%时,产物产率几乎没有受到影响(表2,Entry 4);而当催化剂用量为3 ×10-3mol%时,产物产率明显降低,只有78%(表2,Entry 5),因此将催化剂NiCl2(PPh3)2的用量定为4 ×10-3mol%。为了进一步降低成本,尝试使用无水NiCl2和PPh3配体配合使用来代替NiCl2(PPh3)2,结果发现将该催化剂NiCl2用量由3 ×10-3mol%逐渐增至10-2mol%,产物产率都不理想,仅有80%(表2,Entries 6-9)。综上所述4a 与5a 的摩尔比是1 ∶2,催化剂为NiCl2(PPh3)2且其用量为4 ×10-3mol%。

表1 2-羟基-4-甲氧基甲醚基苯乙酮的合成条件优化aTable 1 Optimization of synthesis of 1-(2-Hydroxy-4-(methoxymethoxy)phenyl)ethanone

表2 镍催化Negishi 交叉偶联反应的条件优化aTable 2 Optimization of synthesis of isoflavone by the Negishi cross-coupling under nickelcatalyst
3 小结
以取代2-羟基苯乙酮和取代溴苯为起始原料,首先合成取代3-碘色原酮,再与芳基锌试剂进行Negishi 交叉偶联反应合成了芒柄花素(7a)、大豆苷元(7b)和鹰嘴豆芽素A(7c)、染料木素(7d)四种药用异黄酮。反应中使用Co(II)催化法制备芳基锌试剂,方法简便,和其它有机金属催化偶联反应合成异黄酮的方法比较,所使用的镍催化剂NiCl2(PPh3)2廉价易得;芳基锌试剂的制备、取代3-碘色原酮的制备和Negishi 交叉偶联反应都是在室温下进行的,全合成反应不涉及高温,不需要惰性气体保护,操作简便、后处理简单,反应条件温和,对环境友好,产品产率高,具有潜在的应用价值。
1 Blank VC,Poli C,Marder M,et al.Antiproliferative activity of various flavonoids and related compounds:additive effect of interferon-α2b.Med Chem Lett,2004,14:133-136.
2 Laupattarakasem P,Houghton PJ,Robin J,et al.Antiinflammatory isoflavonoids from the stems of Derris scandens.Planta Med,2004,70:496-501.
3 Damrongkiet A,Jisnuson S,Prasat K,et al.Antiviral isoflavonoid sulfate and steroidal glycosides from the fruits of Solanum torvum.Phytochemistry,2002,59:459-463.
4 Kang KA,Zhang R,Piao MJ,et al.Protective effect of irisolidone,a metabolite of kakkalide,against hydrogen peroxide induced cell damage via antioxidant effect.Bioorg Med Chem,2008,16:1133-1141.
5 Middleton E,Kandaswami C,Theoharides TC.The effects of plant flavonoids on mammalian cells:Implications for inflammation,heart disease,and cancer.Pharmacol Rev,2000,52:673-751.
6 Balasubramanian S,Nair MG.An efficient“One Pot”synthesis of isoflavones.Synth Commun,2000,30:469-484.
7 Zhang Q,Botting NP.The synthesis of [2,3,4-13C3]glycitein.Tetrahedron,2004,60:12211-12216.
8 Yokoe I,Sugita Y,Shirataki Y.Facile synthesis of isoflavones by the cross-coupling reaction of 3-iodochromone with arylboronic acids.ChemPharm Bull,1989,37:529-530.
9 Vasselin DA,Westwell AD,Matthews CS,et al.Structural studies on bioactive compounds:synthesis and biological properties of fluoro-,methoxyl-,and amino-substituted 3-phenyl-4H-1-benzopyran-4-ones and a comparison of their antitumor activities with the activities of related 2-phenylbenzothiazoles.J Med Chem,2006,49:3973-3981.
10 Rao MLN,Venkatesh V,Jadhav DN.Pd-Catalyzed efficient cross-ouplings of 3-iodochromones with triaryl bismuths as substoichiometric multicoupling organometallic nucleophiles.Synlett,2009,16:2597-2600.
11 Paguette LA,Stucki H.A new general approach to the synthesis of oxygen-containing heterocycles by virtue of hydroxyl neighboring group participation.The condensation of enamines with salicylaldehydes.J Org Chem,1996,31:1232-1235.
12 Ng LT,Ko HH,Lu TM.Potential antioxidants and tyrosinase inhibitors from synthetic polyphenolic deoxybenzoins.Bioorg Med Chem,2009,17:4360-4366.
13 Washburn WN.Preparation of N-[3-(2-aralkylamino-1-hydroxyethyl)phenyl]methanesulfonamides and analogs as β3 adrenoceptor agonists.US5776983,1998.
14 Lee JJ,Kim CB,Park SM,et al.Synthesis of isoflavonoids derivatives via synthetic method involving simplified process and formation of (phenyl)acetophenone intermediates via Fries rearrangement reaction.KR2005076282A,2005.
15 Baker W,Ollis WD.A new synthesis of isoflavones.Nature,1952,169:706.
16 Wahala K,Hase TA.Expedient synthesis of polyhydroxyisoflavones.J Chem Soc Perkin Trans,1991,1:3005-3008.
17 Balasubramanian S,Ward DL,Nair MG.The first isolation and crystal structure of a boron difluoro complex (isoflavone yellow).Biologically active intermediates produced during isoflavone synthesis.J Chem Soc Perkin Trans,2000,1:567-569.
18 Krishnamurty HG,Prasad JS.Synthesis and biological activityof hydroxamic acid derived vasopeptidase inhibitor analogues.Tetrahedron Lett,1977,35:3071-3072.
19 Farkas L,Gottsegen A.Synthesis of sophorol,villanone,lonchoearpan,claussequinone,philenopteran,leiocalycin and some other natural isoflavonoids by the oxidative rearrangement of chaleones with thallium(III)nitrate.J Chem Soc Perkin Trans,1974,1:305-312.
20 Rani I.Two syntheses of 7,2'-dimethoxyisoflavone.Indian J Chem,1987,26B:361-362.
21 Singh OV,Kapil RS.A general method for the synthesis of isoflavones by oxldativerearrangement of flavanones using thallium(III)perchlorate.Indian J Chem,1993,32B:911-915.
22 Zhang ZT,Liang B,Xue D,et al.The synthesis and still cross-coupling reaction of isoflavones.CN102241657.2011.
23 Thomsen,Ib Torssell,Kurt BG.Use of nitrile oxides in synthesis.Novel synthesis of chalcones,flavanones,flavones and isoflavones.Organ Chem Biochem,1988,B42:303-308.
24 Zhang ZT,Qiao JF,Wang D,et al.Synthesis of isoflavones by room-temperature nickel-catalyzed cross-couplings of 3-iodo(bromo)chromones with arylzincs.Mol Divers,2014,18:245-251.
25 Kazmierski I,Gosmini C,Paris JM,et al.New progress in the cobalt-catalysed synthesis of aromatic organozinc compounds by reduction of aromatic halides by zinc dust.Tetrahedron Lett,2003,44:6417-6420.
26 Krasovskiy A,Knochel P.Convenient titration method for organometallic zinc,magnesium and lanthanide reagents.Synthesis,2006,5:890-891.
27 Denis JD,Gordon JS,Carroll VM,et al.Novel synthesis of the isoflavone genistein.Synthesis,2010,10:1590-1592.
28 Yu DL,Chen CH,Brossi A,et al.Anti-AIDS agents 60.Substituted 3'R,4'R-di-O-(-)-camphanoyl-2',2'-dimethyldihydropyrano[2,3-f]chromone(DCP)analogues as potent anti-HIV agents.J Med Chem,2004,47:4072-4082.
猜你喜欢
中南民族大学学报(自然科学版)(2022年6期)2022-11-02
分子催化(2022年1期)2022-11-02
井冈山大学学报(自然科学版)(2021年1期)2021-03-05
中南民族大学学报(自然科学版)(2020年6期)2020-12-22
中国组织化学与细胞化学杂志(2017年1期)2017-06-15
中小学实验与装备(2016年1期)2016-04-19
中国塑料(2015年10期)2015-10-14
中国塑料(2015年8期)2015-10-14
红领巾·探索(2014年8期)2014-10-10
中国洗涤用品工业(2012年4期)2012-03-20
