
焙烧温度对CuO/Co3 O4-CeO2催化剂CO优先氧化性能的影响
2015-02-03 01:54陈业娜
化学工业与工程 2015年2期
陈业娜,孟 明
(天津大学化工学院,天津市应用催化科学与工程重点实验室,天津300072)
近年来,以氢气为燃料的质子交换膜燃料电池(PEMFC)由于具有体积小、可操作温度低、能量密度高以及启动快等优点而获得了快速发展[1-2]。目前质子交换膜燃料电池多以纯氢作为燃料,氢气的主要来源是碳氢化合物的水蒸气在线重整,重整气经过水汽变换的初步处理后,得到的混合气含有体积分数为40% ~75%的 H2,20% ~25%的 CO2,少量的H2O和0.5%~2.0%的CO[3-5],在燃料电池可工作的温度范围内(80~120℃)[6],CO极易吸附在燃料电池的阳极Pt催化剂上,造成其中毒,从而导致PEMFC性能的大幅度下降。研究表明,当CO的浓度降低到10×10-6[7]以下时,燃料电池才能够高效工作。
目前CO的消除有多种方法,分为物理方法和化学方法[8-9]。其中CO优先氧化(CO PROX)的方法是通过催化剂将CO转化成CO2而消除,同时最大限度地降低H2的损耗,该方法被认为是消除富氢气氛中少量 CO最经济和最有效的方法[10-13]。CO优先氧化技术的核心是催化剂,因而研发高性能CO优先氧化催化剂是十分必要的。
研究者们多年的研究发现,铜基催化剂对CO优先氧化具有良好的活性,且催化剂成本低,但催化剂的性能与焙烧温度密切相关[14-15]。我们的研究发现,CuO/Co3O4-CeO2三元氧化物催化剂对 CO优先氧化具有很高的催化性能,尤其是 n(Ce)/n(Ce+Co)为 0.1的载体负载的 CuO催化剂,该比例的载体具有最大的比表面积(500℃焙烧4 h,SBET为 79 m2/g),而 n(Ce)/n(Ce+Co)为 0.3 和 0.5 的载体的比表面积分别只有44和29 m2/g。到目前为止,关于焙烧温度对 CuO/Co3O4-CeO2催化剂活性的影响尚没有任何研究报导。故本研究采用共沉淀和浸渍两步法制备了不同温度焙烧的CuO/Co3O4-CeO2三元氧化物催化剂,考察了它们对CO的优先氧化性能及活性稳定性,优化了催化剂的焙烧温度,应用XRD、BET、H2-TPR和 XAFS等技术对催化剂的结构进行了详细表征,并探讨了催化剂的结构与性能的关系。
1 实验部分
1.1 载体和催化剂的制备
首先采用共沉淀法制备了 n(Ce)/n(Ce+Co)为0.1的Co3O4-CeO2载体(CoCe10)。具体步骤如下:按化学计量比例配置适量的 Ce(NO3)3·6H2O和 Co(NO3)2·6H2O混合溶液,在机械搅拌条件下逐滴加入事先配置好的2 mol/L的NaOH溶液,直至pH值达到12.0,室温下继续搅拌1 h,然后移入60℃水浴中老化2 h,将所得到的沉淀液进行过滤,并用热蒸馏水洗涤数次,得到的沉淀于120℃烘箱中干燥12 h后,再于500℃焙烧4 h,制得最终的载体。
CuO的负载采用硝酸盐水溶液等量浸渍法,其负载量选择质量分数7%[16],这主要是根据制得的载体的比表面积来确定的,因为负载量太大,CuO的分散度会降低,而负载量太小,表面CuO组分会减少,这些对催化活性都不利。催化剂的具体制备步骤如下:取已知质量的载体粉末浸渍于适量的Cu(NO3)2·3H2O溶 液 中,并 在 超 声 条 件 下 浸 渍20 min,然后在室温下静置24 h,在120℃烘箱中干燥12 h。将干燥后的样品平均分为5份,分别在250、300、350、400 和 450 ℃焙烧 4 h,制得不同温度焙烧的催化剂,记为 CuCoCe-X,其中 X表示焙烧温度。
1.2 催化剂表征
1.2.1 比表面积(BET)测试
催化剂的比表面积测试(BET)是在美国康塔公司生产的 Quantachrome QuadraSorb SI物理吸附仪上进行的,在液氮温度(77 K)下进行N2的吸附/脱附实验。样品在测试前预先在300℃真空脱气处理3 h。用BET吸附曲线的线性部分(p/p0=0.089~0.297)来计算样品的比表面积(SBET)。
1.2.2 X射线粉末衍射(XRD)测试
X射线粉末衍射测试(XRD)是在 Rigaku D/Max-2500型号的多晶粉末衍射仪上进行的,Cu-Kα(λ=0.15418 nm)作为射线源。管电压为40 kV,电流为20 mA,采集2θ从10°到 90°范围内的数据,采集步长为0.02°。
1.2.3 X-射线吸收精细结构测定(XAFS)
X-射线吸收精细结构(XAFS)的测定在北京同步辐射实验室的BSRF 1W1B光束线上的XAFS站进行。储存环电子能量约为2.2 GeV,平均环电流为150 mA,采用硅(111)双晶单色器获取单色光。标样中的Cu元素的K边XAFS谱采用透射模式采集,催化剂样品中的Cu元素的K边XAFS谱采用荧光模式采集。利用Athena软件进行数据处理,获得Cu的K边径向结构函数(RSF),包括背景扣除,归一化处理,μ0拟合和Fourier变换。
1.2.4 程序升温还原(H 2-TPR)测试
程序升温还原(H2-TPR)实验是在天津先权仪器公司生产的TP-5079 TPDRO仪器上进行,采用热导检测器。反应气为8%的H2/N2混合气,催化剂用量为30 mg,气体流速为30 mL/min,以10℃/min的速率从室温升到600℃。反应气在进入检测器之前,先用 CaO+NaOH材料除去气氛中的 H2O和CO2。
1.3 催化剂活性评价
催化剂的活性评价是在内径为8 mm的石英管式固定床反应器中进行,石英管长约50 cm,40~60目的催化剂(500 mg)置于反应管内恒温区的石英棉上。原料气组成(体积分数)为1%CO,1%O2,50%H2,N2作为平衡气。空速约12 000 h-1。反应原料气和产物采用Agilent 7890A型色谱在线分析,采用 TCD检测器。通过式(1)、(2)计算CO的转化率(X)及氧气对 CO2的选择性(S)。

其中,CCOin,CCOout,CO2in,CO2out分别表示 CO和氧气在进口处和出口处的浓度。
2 实验结果与讨论
2.1 催化剂的CO PROX活性
对经过不同温度焙烧的CuCoCe10催化剂在富氢条件下的CO优先氧化性能进行了考察,结果如图 1 所示,V(H2)/V(CO)/V(O2)/V(H2O)/V(CO2) =50/1/1/0/0,N2为平衡气。
从图1a)可以看出,随着反应温度的不断升高,不同温度焙烧的CuCoCe-X催化剂对CO的转化率都迅速提高,其中,350℃焙烧的CuCoCe10催化剂具有最好的 CO优先氧化活性,在98℃时就能将CO完全转化,而对于其它温度250、300和450℃焙烧的催化剂分别在160、156和177℃使 CO完全转化,在400℃焙烧的催化剂并没有能够使CO的转化率达到100%。同时,在350℃焙烧的催化剂也具有最宽的CO完全转化窗口,具体的活性数据列于表1中。
从图1b)可以发现,在100℃之前,所有催化剂上O2到CO2的选择性均保持100%。随反应温度的升高该选择性有所下降,此时,不同温度焙烧的催化剂上O2到CO2的选择性与催化剂对CO的转化率似乎呈现相反的趋势。但考虑到燃料电池阳极催化剂对CO的极其敏感性,认为CO转化率是衡量一个催化剂CO优先氧化性能的最重要标准。因此,综合考虑,350℃焙烧的CuCoCe10催化剂对CO
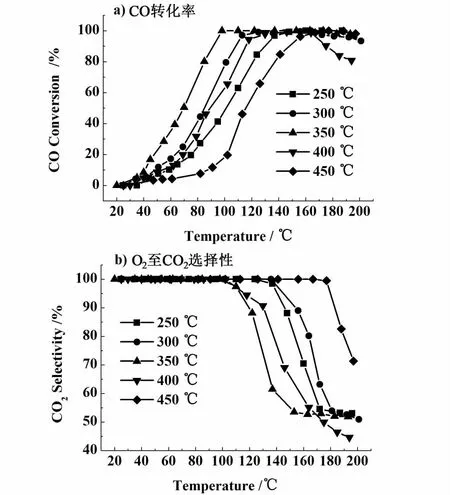
图1 CuCoCe-X催化剂的CO PROX活性图Fig.1 Activity curves of the CuCoCe-X catalysts for CO p referential oxidation
优先氧化反应具有相对最好的催化性能。
2.2 比表面积(BET)结果分析
不同温度焙烧的CuCoCe-X催化剂的比表面积数据列于表1中。从表1中可看到,350℃焙烧的CuCoCe10催化剂具有最大的比表面积(79 m2/g)。当焙烧温度不高于350℃时,焙烧温度的提高有利于硝酸盐前驱体分解为氧化物,同时产生孔结构,因而比表面积随焙烧温度升高而升高;在350℃以上,由于硝酸盐已基本完全分解,因而继续提高焙烧温度,必然导致催化剂的烧结,故比表面积呈现明显的下降趋势。催化剂的比表面积大小顺序基本上与它们的CO PROX反应活性一致,表明Cu物种在催化剂表面的分散性对活性有直接影响。

表1 CuCoCe-X催化剂的活性数据和比表面积Tab le 1 Specific sur face area’CO half conversion temperature(T50)’ CO fu ll conversion temperature(T100)’ and CO fu ll conversion temperature-window(T W)
2.3 XRD结果分析
图2是不同温度焙烧的CuCoCe-X催化剂的XRD谱图。

图2 CuCoCe-X催化剂的XRD谱图Fig.2 XRD patterns of CuCoCe-X catalysts
从图2中可以看出,不同温度焙烧的CuCoCe10催化剂样品都出现了典型的萤石型CeO2[17]和尖晶石型Co3O4[18]的衍射峰。随着焙烧温度的升高,400和450℃焙烧的样品的XRD谱图上出现了CuO相的衍射峰,说明焙烧温度过高时,高分散状态的CuO物种发生了烧结,使CuO颗粒尺寸增大,这与BET结果一致。这种大颗粒的CuO相的形成对CO优先氧化反应不利,因此高温焙烧的样品的催化性能相对较低。
2.4 XAFS结果分析
由于350℃及更低温度焙烧的催化剂的XRD谱图中没有发现CuO的衍射峰,为了能够确定催化剂中的Cu物种,对不同温度焙烧的催化剂进行了EXAFS测试,图3示出了CuCoCe-X催化剂中Cu的K边径向结构函数图。从图3中可以看出,不同温度焙烧的催化剂的RSFs中出现的主配位峰均与标样CuO的接近,可以确定催化剂样品中的铜物种主要以CuO形式存在。

图3 不同温度焙烧的CuCoCe10催化剂及CuO’Cu2 O和Cu标样的K边径向结构函数图Fig.3 Radial structure functions(RSFs) of Cu K-edge of the reference compounds and catalysts
2.5 H 2-TPR结果分析
为了研究不同温度焙烧的催化剂的氧化还原性质,对催化剂样品进行了H2-TPR实验,结果如图4所示。
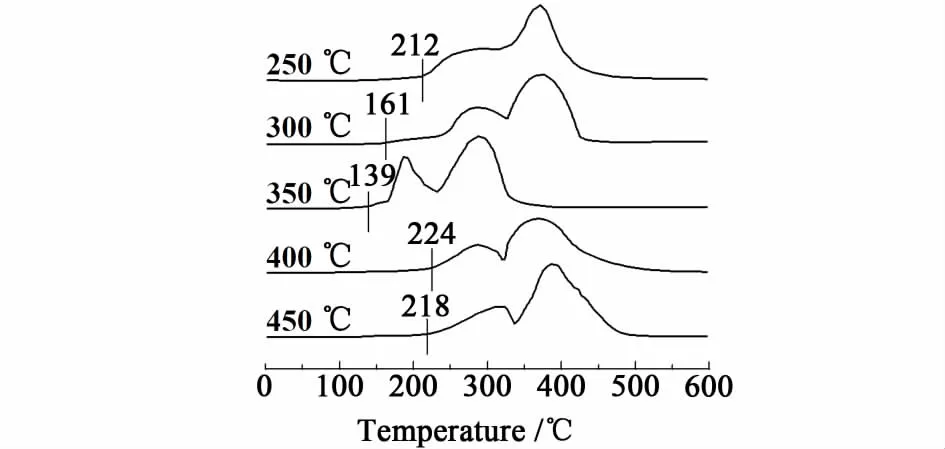
图4 CuCoCe-X催化剂的H 2-TPR谱图Fig.4 H 2-TPR profiles of the CuCoCe-X catalysts
从图4可以看出,不同温度焙烧的 CuCoCe10催化剂的还原峰有所不同。其中,350℃焙烧的催化剂被还原的起始温度最低,约为139℃,而在250、300、400和450℃焙烧的 CuCoCe10催化剂的起始被还原温度分别为212、161、224和218℃。由此可见,在350℃焙烧的CuCoCe10催化剂具有最好的氧化还原(Redox)性能。H2-TPR的结果与催化剂的CO PROX反应活性相一致。
综上所述,350℃是催化剂的最适宜焙烧温度,焙烧温度过高或过低都对CuO和载体之间的相互作用产生不利影响,从而影响 CuCoCe10催化剂最终的CO PROX反应活性。
2.6 稳定性测试
在CO PROX反应中,催化剂的寿命也是影响其应用的一个重要方面。因此,我们对350℃焙烧的、活性最好的CuCoCe10催化剂进行了稳定性考察,空速设定为12 000 mL/h/g,反应温度为120℃。测试结果如图5。在反应进行后的前20 h,采用的反应气为体积分数 1%CO,1%O2,50%H2,N2作为平衡气,而经过20 h的CO PROX反应后,在反应气中添加了10%的 CO2,继续考察催化剂的活性,经30 h后,再添加12.5% 的H2O蒸汽,继续反应。经过85 h的稳定性测试后,CO的转化率仅由刚开始的100%下降到91%,O2至CO2的选择性也只下降了15%,可见,CuCoCe10催化剂具有较高的活性稳定性。
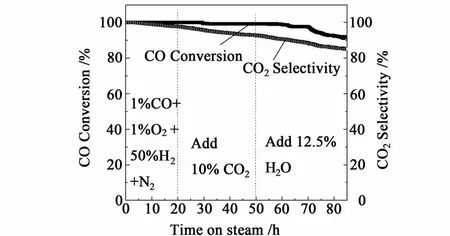
图5 最优催化剂CuCoCe10-350在120℃的稳定性测试Fig.5 The stability test over the op timal catalyst CuCoCe10-350 at 120℃
从图5还可以看到,H2O和CO2的存在对催化剂在的CO优先氧化反应性能产生了一定的不利影响,这与文献报道的相同[19-20]。分析认为,CO2的存在可能与CO发生了竞争吸附,并在催化剂表面生成了表面碳酸盐,导致CO优先氧化反应活性的下降[16]。H2O的存在会引起催化剂上CO活性中心的堵塞,或者形成CO-H2O复合物种,抑制CO的活化,使其不能被O2氧化[17-18],从而最终导致催化剂对CO优先氧化反应活性的下降。
3 结论
经过不同温度焙烧后的催化剂样品中,铜物种主要是以CuO相存在的。350℃焙烧的CuCoCe10催化剂,具有最大的比表面积和最好的氧化还原性能,能够促进铜物种的分散及其与载体的相互作用。活性测试结果表明,该催化剂在98~173℃范围内都能将CO完全转化为CO2,具有最好的CO优先氧化反应性能和最宽的可操作温度窗口。当催化剂的焙烧温度过低或过高时,CuO与载体间的相互作用太弱或太强,不利于CuO活性相对CO的优先氧化。对CuCoCe10-350进行85 h的活性稳定性测试后发现,虽然CO转化率和O2至CO2的选择性略有下降,但该催化剂仍表现出较高的活性稳定性;反应气中H2O和CO2的存在不利于CO的优先氧化,原因可能是水分子的吸附堵塞了CO氧化活性中心,或CO2的吸附在活性中心上形成了不利于反应的表面碳酸盐物种。
[1]Bose S,Kuila T.Polymermembranes for high temperature proton exchange membrane fuel cell:Recent advances and challenges[J].Prog PolymSci,2011(36): 813-843
[2]Wee J.Applications of proton exchange membrane fuel cell systems[J].RenewSust Energ Rev, 2007 (11):1 720-1 738
[3]Avgouropoulos G,Manzoli M,Boccuzzi F,et al.Catalytic performance and characterization of Au/doped-ceria catalysts for the preferential CO oxidation reaction[J].J Catal, 2008, 256(2): 237-247
[4]Pozdnyakova O,Teschner D,Wootsch A,et al.Preferential CO oxidation in hydrogen(PROX) on ceria-supported catalysts, part I: Oxidation state and surface species on Pt/CeO2under reaction conditions[J].JCatal,2006, 237(1):1-16
[5]Snytnikov P V,Sobyanin V A,Belyaev V D,et al.Selective oxidation of carbon monoxide in excess hydrogen over Pt-,Ru-and Pd-supported catalysts[J].Appl Catal A, 2003, 239(1/2): 149-156
[6]Zepeda T, Martinez-Hernández A, Guil-López R, et al.Preferential CO oxidation in excess of hydrogen over Au/HMS catalysts modified by Ce,Fe and Ti oxides[J].Appl Catal B, 2010, 100(3/4): 450-462
[7]Choudhary T,Goodman D.CO-Free fuel processing for fuel cell applications[J].Catal Today, 2002, 77(1/2): 65-78
[8]严菁,马建新,万钢.用于燃料电池汽车重整器的CO脱除技术[J].天然气化工,2002,27:35-38 Yan Jing, Ma Jianxin, Wan Gang.Methods of CO2clean up for the reformer of fuel cell vehicles[J].Natural Gas Chemical Industry, 2002, 27: 35-38(in Chinese)
[9]刘娅琼,吕倩,孟明.制备参数对Au/TiO2催化剂上CO低温氧化性能的影响[J].化学工业与工程,2013, 30(5): 1-6 Liu Yaqiong, Lyu Qian, Meng Ming.Effect of preparation parameters on the CO low-temperature oxidation performance of Au/TiO2catalysts[J].Chemical Industry and Engineering, 2013, 30(5): 1-6(in Chinese)
[10]Bion N,Epron F,Moreno M.Preferential oxidation of carbon monoxide in the presence of hydrogen (PROX)over noble metals and transition metal oxides:Advantages and drawbacks[J].Top Catal, 2008, 5(1/4):76-88
[11]余强,高飞,董林.铜基催化剂用于一氧化碳催化消除研究进展[J].催化学报,2012,33(8):1 245-1 256 Yu Qiang, Gao Fei, Dong Lin.Recent progress of Cubased catalysts for catalytic elimination of CO[J].Chinese Journal of Catalysis, 2012, 33(8): 1 245-1 256(in Chinese)
[12]Chen Y,Lee D S,Chen H.Preferential oxidation of CO in H2streamon Au/ZnO-TiO2catalysts[J].Int J Hydrogen Energy, 2012, 37(20): 15 140-15 155
[13]Meng M,Liu Y,Sun Z,et al.Synthesis of highly-dispersed CuO-CeO2catalyst through a chemisorption-hydrolysis route for CO preferential oxidation in H2-rich stream[J].Int J Hydrogen Energy 2012; 37(19):14 133-14 142
[14]Moretti E,Storaro L,Talon A,et al.Effect of thermal treatments on the catalytic behaviour in the CO preferential oxidation of a CuO-CeO2-ZrO2catalyst with a flowerlikemorphology[J].Appl Catal B,2011,102 (3/4):627-637
[15]Tu Y,Meng M,Sun Z,et al.CO preferential oxidation over Au/MnOx-CeO2catalysts prepared with ultrasonic assistance:Effect of calcination temperature[J].Fuel Process Technol, 2012, 93(1): 78-84
[16]Cao J,Wang Y,Sun G,et al.CuO/CexSn1-xO2catalysts: Synthesis, characterization, and catalytic performance for low-temperature CO oxidation[J].Transition Met Chem,2011,36(1):107-112
[17]Gurbania A, Ayastuya J L, González-Marcos MP, et al.Comparative study of CuO-CeO2catalysts prepared by wet impregnation and deposition-precipitation[J].Int JHydrogen Energy, 2009, 34(1): 547-553
[18]Gómez L E,Tiscornia IS,Boix A V,et al.Co/ZrO2catalysts coated on cordieritemonoliths for CO preferential oxidation[J].Appl Catal A, 2011, 401(1/2): 124-133
[19]Park JW,Jeong JH,Yoon WL,et al.Selective oxidation of carbon monoxide in hydrogen-rich streamover Cu-Ce/γ-Al2O3catalysts promoted with cobalt in a fuel processor for proton exchangemembrane fuel cells[J].J Power Sources, 2004, 132(1/2): 18-28
[20]Gamarra D, Martínez-Arias A.Preferential oxidation of CO in rich H2over CuO/CeO2:Operando-DRIFTS analysis of deactivating effect of CO2and H2O[J].JCatal,2009, 263(1): 189-195
猜你喜欢
华人时刊(2022年9期)2022-09-06
小学生学习指导(高年级)(2022年3期)2022-03-29
华人时刊(2020年15期)2020-12-14
商周刊(2018年25期)2019-01-08
传媒评论(2018年5期)2018-07-09
小学生导刊(高年级)(2017年2期)2017-06-10
小学生导刊(2017年6期)2017-02-10
中国卫生(2016年12期)2016-11-23
小学生导刊(高年级)(2016年1期)2016-01-29
广州大学学报(自然科学版)(2015年4期)2015-12-23
