
磷酸酯类单体与氧化锆间化学反应模型的建立及作用机制
2017-05-04 00:35卢枳岑谢海峰章非敏张怀勤陈晨
华西口腔医学杂志 2017年2期
卢枳岑 谢海峰 章非敏 张怀勤 陈晨
1.南京医科大学口腔医院修复科;2.南京医科大学口腔疾病研究江苏省重点实验室;3.南京医科大学口腔医院牙体牙髓科,南京 210029
磷酸酯类单体与氧化锆间化学反应模型的建立及作用机制
卢枳岑1,2谢海峰1,2章非敏1,2张怀勤1,2陈晨2,3
1.南京医科大学口腔医院修复科;2.南京医科大学口腔疾病研究江苏省重点实验室;3.南京医科大学口腔医院牙体牙髓科,南京 210029
目的 对磷酸酯单体10-甲基丙烯酰氧癸二氢磷酸酯(MDP)提高四方相氧化锆与树脂间化学粘接的作用机制进行解释。方法分别构建MDP、四方相氧化锆陶瓷晶体以及两者反应的数字模型,采用热力学方法对MDP分子与四方相氧化锆晶体簇相互作用的吉布斯自由能和平衡常数进行计算。结果MDP与四方相氧化锆晶体簇可能发生单配位形式和双配位形式的结合,其吉布斯自由能分别为-147.761和-158.073 kJ·mol-1,说明MDP能够与四方相氧化锆晶体发生化学结合,平衡常数较高(4.95×1027)的双配位结合形式较平衡常数较低(7.72×1025)的单配位结合形式更为稳定。结论MDP能够通过与四方相氧化锆陶瓷以双配位形式发生化学结合来提高其与树脂的粘接强度。
量子化学; 反应机制; 氧化锆; 粘接; 磷酸酯单体; 底涂剂
氧化钇稳定四方相氧化锆(Yttria-stabilized tetragonal zirconia polycrystals,Y-TZP)陶瓷是口腔冠、桥修复体制作最常用的材料之一。由于Y-TZP陶瓷化学惰性高,硅烷化等常规提高陶瓷粘接的方法对其无效[1-3]。磷酸酯单体10-甲基丙烯酰氧癸二氢磷酸酯(10-methacryloyloxydecyl dihydrogen phosphate,MDP)的问世实现了通过简单的操作来提高Y-TZP陶瓷粘接的目的,因此获得了临床上的极大关注。尽管当前普遍认为MDP之所以能与特殊性质的氧化锆表面形成化学结合是因为其中磷酸基团的作用[3-5],然而目前仍无法直接从理论上解释其具体机制。对于机制了解的缺乏极大地限制了具有更好性能的磷酸酯单体的开发。对于解决这样的问题,量子化学方法具有先天的优势,其可在电子水平上对不同分子间形成化学键的稳定性进行精细的理论研究,直接论证化学键的稳定性,分析反应过程,这是传统实验表征方法所难以替代和无法实现的。本课题组在之前关于包含MDP的处理剂对牙科氧化锆陶瓷与树脂粘接性能影响的研究中,曾运用量子化学方法评价pH环境在其中发挥的作用[6]。由于类似方法在牙科粘接领域的应用较少,为了介绍该方法,并为今后类似研究提供可以借鉴的实验资料,本研究详细描述通过量子化学的方法模拟氧化锆与MDP结合时界面上的可能化学反应,并对反应机制加以理论阐释。
1 材料和方法
Y-TZP中氧化锆的晶型为四方相,虽然其中掺杂氧化钇作为稳定相,但牙科Y-TZP含量通常介于3%~5%,对四方相氧化锆的晶胞参数改变不大,因此无需考虑掺杂的情况。根据ICSD数据库的晶体结构信息和MDP分子式结构,用Gaussview软件(Gaussian公司,美国)分别构建四方相氧化锆晶体和MDP的数字模型并进行优化。
由于MDP调节Y-TZP是发生在界面的反应,并且实际中四方相氧化锆陶瓷的表面结构可能不完整,因此需要将建立的四方相氧化锆晶体数字模型切割出多种原子排列的氧化锆表面来研究MDP分子与晶面可能的作用位点。切割晶面时人为的暴露锆原子,因为只有锆原子暴露的晶面才可能与MDP分子发生作用。由于直接对氧化锆表面模型进行计算会导致庞大的数据体系而无法实现,但氧化锆是一个周期性晶体,可以通过从确定表面的数字模型切割出一个小的中性重复单元(Zr4O8)作为后续计算用的簇模型。为了能够以切割出的模型代替整体并保证结果的正确性,这个簇模型需要拥有大部分氧化锆晶体的特征,包括:中性金属氧化物簇;大小适中,可以用于采用“从头计算法”;在几何构型优化后,氧化锆簇构型基本稳定。
关于计算配体与金属氧化物的相互作用有数种算法。当前实验中,MDP分子与Zr4O8簇的相互作用使用分层计算(our own N-layered integrated molecular orbital+molecular mechanics,ONIOM)方法进行研究,此种方法可以处理较大体系的运算。密度泛函理论(density functional theory,DFT)算法和分子动力学(molecular mechanics,MM)算法分别用来处理高精度和低精度计算。DFT算法使用了B3LYP密度泛函优化MDP分子磷酸基团的几何结构和计算其热力学数据。碳、磷、氧和氢原子使用了6-311+ G(d,p)基组,其中碳、磷、氧和氢原子都加上极化函数,而只有碳、磷、氧原子加上弥散函数。对于电子数较多的锆原子,LanL2DZ基组描述价层电子和内层电子以及相关的相对论效应赝势。MM算法使用了UFF力场来进行脂链和含双键尾基的几何构型优化和热力学数据的计算。因为几何优化可能会改变初始结构,所以本研究在优化时固定氧化锆单元的位置来最大程度确保四方相氧化锆晶体表面的特性。溶剂效应使用积分方程极化连续介质模型(integral equation formal polarization continuum model,IEF-PCM)。以上所有计算均通过Gaussian 09软件(Gaussian公司,美国)完成。
2 结果
图1和2分别为建立的MDP分子与四方相氧化锆晶体模型。根据模型,MDP分子的磷酸基团中,两个羟基氧的距离大约为2.5 Å(1 Å=100 nm),氧化锆晶面上锆原子的距离3.6~6.4 Å不等,氧化锆晶面上的锆原子距离决定了MDP分子与氧化锆晶体形成配位键可能通过“双配位”或“单配位”两种反应通道实现(图3)。当锆原子之间的距离较小时,磷酸基团上的两个羟基氧的孤对电子轨道与锆原子上的空轨道有效重叠,形成双配位,反之则形成单配位。图4为优化后的符合计算要求的中性重复单元金属氧化物簇Zr4O8簇模型,优化后的MDP分子在Zr4O8晶体簇表面吸附的模型见图5。

图1 建立的MDP分子模型Fig1 The model of MDP molecule
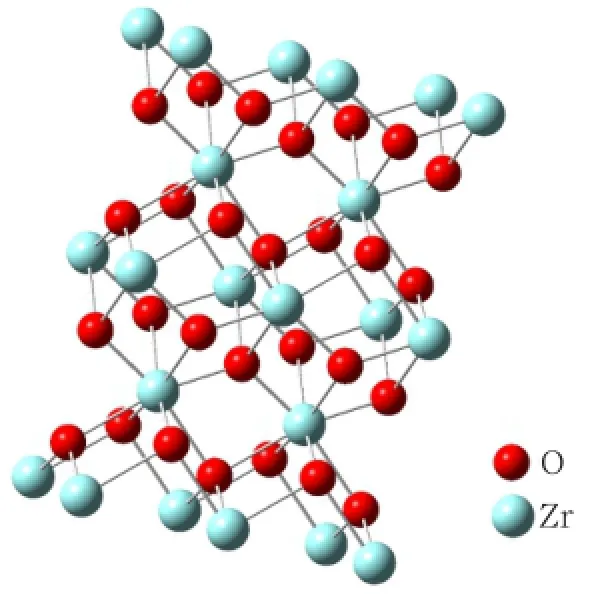
图2 建立的四方相氧化锆晶体模型Fig2 The model of tetragonal zirconium dioxide crystals

图3 MDP分子与氧化锆晶体可能形成的配位键形式(优化前)Fig3 Possible coordinate configurations between MDP and zirconium dioxide (before optimization)

图4 优化后的符合计算要求的中性重复单元金属氧化物簇Zr4O8簇模型Fig4 The model of Zr4O8cluster structure after optimization by Gaussian
根据可能的两条反应通道,MDP与Zr4O8晶体簇作用的化学反应过程如下方两个公式所示,解离为公式1,配位为公式2。其中,R为MDP分子中除磷酸基团以外的部分,OP(OH)2为MDP的磷酸基团,aq为溶液,两种配位方式都适用公式2。

对以上方程的吉布斯自由能和反应平衡常数的计算结果见表1~3。

图5 优化后的MDP分子在Zr4O8晶体簇表面吸附的模型Fig5 The model of Zr4O8cluster attached with MDP molecule after optimization

表1 反应通路公式1的热力学数据Tab1 Thermodynamic data of formula 1

表2 反应通路公式2的热力学数据(双配位形式)Tab2 Thermodynamic data of formula 2 (double coordinate configuration)

表3 反应通路公式2的热力学数据(单配位形式)Tab3 Thermodynamic data of formula 2 (single coordinate configuration)
综合公式(1)和(2)可以看出,MDP与Zr4O8晶体簇发生双配位结合反应的综合吉布斯自由能ΔG= ΔG1+ΔG2=-158.073 kJ·mol-1,而发生单配位形式结合的吉布斯自由能ΔG=ΔG1+ΔG3=-147.761 kJ·mol-1,这两种反应途径吉布斯自由能数值前的负号说明反应均能够正向自发发生。发生单配位形式结合的平衡常数7.72×1025小于发生双配位结合形式的平衡常数4.95×1027,说明双配位比单配位形式更为稳定。
3 讨论
MDP是一种双性功能分子,其调节Y-TZP陶瓷与树脂粘结性能的作用类似于硅烷在增强玻璃基陶瓷与树脂粘接性能中所扮演的角色。通常认为,MDP一端的磷酸基团可与氧化锆表面的“O-Zr-O”之间形成共价键,而另一端的“C=C”可与聚合前树脂水门汀基质中的“C=C”发生加聚反应,从而达到提高氧化锆陶瓷与树脂粘接性能的目的[3-5]。多年来陆续有研究[7-9]利用红外光谱、X线光电子能谱分析等多种界面的表征分析手段来分析Y-TZP陶瓷表面经过MDP调节后的化学键变化,然而都未能直接证明新生成的化学键到底是什么,一直到2012年,Chen等[10]通过飞行时间二次离子质谱首次发现了包含MDP的涂底剂Z-Prime Plus(Bisco公司,美国)与Y-TZP陶瓷表面接触后出现“P-O-Zr”键,支持了MDP中应是磷酸基团能够与氧化锆发生化学反应的推测。然而,尚未有其他实验能够直接检测到该化学键的形成,也没有其他包含MDP的产品被检测出能够与氧化锆陶瓷间形成该键,因此,单纯依靠这一报道无法证实MDP与氧化锆间确实发生了预想的反应。另外,常温下磷酸并不会与Y-TZP陶瓷发生反应[11],这说明脱离MDP整体分子结构时,仅依靠磷酸基团是不具备调节Y-TZP陶瓷的化学活性的。可见,MDP分子中的其他结构对于磷酸基团活性的发挥也具有不可缺少的作用。以上问题显然单纯依靠仪器检测反应产物是难以解释的,必须予以理论证实。明确磷酸酯单体中分子结构影响磷酸基团的化学活性的方式有助于设计和更好地改进磷酸酯单体的分子结构,使其在临床上发挥最佳性能,对于磷酸酯单体的研发具有十分重要的理论意义,因而了解磷酸酯单体与氧化锆陶瓷最基本的反应机理是必要的。
基于量子化学的原理,可以通过模拟各个原子之间的距离与周围的电子来判断原子间成键与否,并通过以基态密度为变量,将体系能量最小化之后得到基态能量。本研究采用了量子化学方法对MDP与四方相氧化锆陶瓷化学反应的发生的可能性及如果反应发生的条件下所形成的化学键进行了解释和分析。锆原子有空电子轨道,MDP分子磷酸基团中的羟基氧有孤对电子,这是二者产生配位连接的充分条件。MDP中存在的末端磷酸基团可以与一个或多个锆原子结合,而这取决于晶体表面锆原子的距离大小。如果晶面几个锆原子距离适当,配位则较为稳定,反应可能发生;反之,如果几个锆原子距离过大,此时氧原子就不可能与多个锆原子发生结合。根据建立的四方相氧化锆晶体表面锆原子间的距离和MDP分子的数字模型可以初步判断,MDP分子只可能采取“双配位”或“单配位”形式与氧化锆晶体形成配位键。为了进一步明确究竟这两种配位键中何者更为稳定,即究竟MDP与氧化锆发生化学结合时会以形成哪一种化学键为主,本实验对两种化学反应通道的吉布斯自由能和平衡常数进行了相应计算。根据结果可以发现,对应MDP解离的公式1的平衡常数比单纯的磷酸一二级电离常数小,其原因可能是由于计算的精度偏差和MDP分子中脂链的推电子作用而导致磷原子的电子密度增加,酸性降低,同时,由于计算结果哈特里(Hartree)是一个很大的能量单位,即使很小的差别也会造成能量计算的较大误差,但综合来讲,对于判断当前反应的趋势是准确的,而想要定量的说明反应的强度则需进行进一步计算。公式2在计算时认为是在气固界面反应,因为磷酸基团接近二氧化锆晶面时溶剂化作用不如在液相显著,并且此时疏水的脂链接触液相,因此本实验并没有考虑溶剂化效应,这使得计算的结果比实际磷酸与金属的配位结合常数偏大,如果考虑溶剂化效应,则体系需克服静电力做功,导致吉布斯自由能更正,从而使平衡常数减小,尽管如此,但由于变化幅度很小,因此并不影响计算的结论。
根据热力学计算的最终结果,MDP与氧化锆晶体间双配位形式的化学键较单配位形式更为稳定,说明MDP调节Y-TZP陶瓷表面应以形成双配位为主要形式。在这一作用机制的基础上,今后的研究重点将放在改善磷酸酯单体分子结构或寻找能够增强Zr-O-P键强度的途径上,以获得能够进一步提高YTZP陶瓷与树脂粘接性能的磷酸酯单体。除此以外,所设计的氧化锆晶体模型还可用于与其他粘结剂在接触面的反应的量子化学定性计算,在理论的支持下可以有效简化其他实验的工作量,并提供必要的指导作用。
[1] Kitayama S, Nikaido T, Takahashi R, et al. Effect of primer treatment on bonding of resin cements to zirconia ceramic [J]. Dent Mater, 2010, 26(5):426-432.
[2] Wegner SM, Kern M. Long-term resin bond strength to zirconia ceramic[J]. J Adhes Dent, 2000, 2(2):139-147.
[3] Özcan M, Bernasconi M. Adhesion to zirconia used for dental restorations: a systematic review and meta-analysis[J]. J Adhes Dent, 2015, 17(1):7-26.
[4] De Souza G, Hennig D, Aggarwal A, et al. The use of MDP-based materials for bonding to zirconia[J]. J Prosthet Dent, 2014, 112(4):895-902.
[5] Aboushelib MN, Matinlinna JP, Salameh Z, et al. Innovations in bonding to zirconia-based materials: PartⅠ[J]. Dent Mater, 2008, 24(9):1268-1272.
[6] Xie H, Tay FR, Zhang F, et al. Coupling of 10-methacrylo yloxydecyldihydrogenphosphate to tetragonal zirconia: Effect of pH reaction conditions on coordinate bonding[J]. Dent Mater, 2015, 31(10):e218-e225.
[7] Lung CY, Botelho MG, Heinonen M, et al. Resin zirconia bonding promotion with some novel coupling agents[J]. Dent Mater, 2012, 28(8):863-872.
[8] Papia E, Larsson C, Du T M, et al. Bonding between oxide ceramics and adhesive cement systems: a systematic review [J]. J Biomed Mater Res B Appl Biomater, 2014, 102(2): 395-413.
[9] Thompson JY, Stoner BR, Piascik JR, et al. Adhesion/cementation to zirconia and other non-silicate ceramics: where are we now[J]. Dent Mater, 2011, 27(1):71-82.
[10] Chen L, Suh BI, Brown D, et al. Bonding of primed zirconia ceramics: evidence of chemical bonding and improved bond strengths[J]. Am J Dent, 2012, 25(2):103-108.
[11] Blatz MB, Sadan A, Kern M. Resin-ceramic bonding: a review of the literature[J]. J Prosthet Dent, 2003, 89(3):268-274.
(本文编辑 李彩)
Establishment and mechanisms of chemical interaction between phosphate monomer and zirconia model
Lu Zhicen1,2, Xie Haifeng1,2, Zhang Feimin1,2, Zhang Huaiqin1,2, Chen Chen2,3. (1. Dept. of Prosthetics, Affiliated Hospital of Stomatology, Nanjing Medical University, Nanjing 210029, China; 2. Jiangsu Key Laboratory of Oral Diseases, Nanjing Medical University, Nanjing 210029, China; 3. Dept. of Conservative Dentistry and Endodontics, Affiliated Hospital of Stomatology, Nanjing Medical University, Nanjing 210029, China)
Objective To analyze chemical mechanism of bonding improvement of zirconia via 10-methacryloyloxydecyl dihydrogen phosphate (MDP) conditioning.MethodsVarious models were created for tetragonal zirconia crystals, molecular MDP, and MDP complex, and tetragonal zirconia crystal. Thermodynamic methods were used to analyze configuration between MDP and tetragonal zirconia crystal through calculation of their Gibbs free energy values and equilibrium constants. Results Two potential configurations (double- and single-coordinate) may occur between MDP and ZrO2crystal clusters. Thermodynamic calculations showed that -147.761 and -158.073 kJ·mol-1Gibbs free energy were required to form single- and double-coordinate configurations; their negative signs indicate that reactions for both configurations can occur. Equilibrium constant for singlecoordinate configuration was 7.72×1025, which was less than that of double-coordinate configuration (4.95×1027), suggesting that the latter was more stable.ConclusionMDP can spontaneously establish a double-coordinate configuration with zirconia.
quantum chemistry; reaction mechanism; zirconia; bonding; phosphate monomer; primer
R 783.1
A
10.7518/hxkq.2017.02.007
Supported by: The National Natural Science Foundation of China (81400539); Priority Academic Program Development of Jiangsu Higher Education Institutions (2014-37); Natural Science Foundation of Jiangsu Province (BK20150998, BK20140913); Natural Science Foundation for Jiangsu Higher Education Institutions (15KJB320003). Correspondence: Chen Chen, E-mail: ccchicy@njmu.edu.cn.
2016-07-16;
2016-10-8
国家自然科学基金(81400539);江苏高校优势学科建设工程资助项目(2014-37);江苏省自然科学基金(BK20150998,BK20140913);江苏省高校自然科学基金(15KJB320003)
卢枳岑,硕士,E-mail:fjlzc92@163.com
陈晨,副主任医师,博士,E-mail:ccchicy@njmu.edu. cn
猜你喜欢
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09
当代陕西(2019年6期)2019-04-17
佛山陶瓷(2017年7期)2017-09-06
中华老年口腔医学杂志(2016年5期)2016-03-01
中国粮油学报(2016年1期)2016-02-06
中国资源综合利用(2016年7期)2016-02-03
西南军医(2015年2期)2015-01-22
外语学刊(2014年3期)2014-12-03
中国粮油学报(2014年8期)2014-02-06
郑州大学学报(理学版)(2013年3期)2013-03-11
