
基因组选择及其在作物育种中的应用
2018-01-16 18:38姜淑琴孙炳蕊潘大建范芝兰陈文丰
广东农业科学 2017年9期
陈 雨,姜淑琴,孙炳蕊,潘大建,范芝兰,陈文丰,李 晨
(1.广东省农科院水稻研究所/广东省水稻育种新技术重点实验室,广东 广州 510640;2.中国农业大学农学院,北京 100193)
品种(系)选育是遗传改良的重要手段之一,在人类农业生产中占着重要地位。传统育种主要根据个体表型和个人经验,结合简单的统计方法,对个体进行选择,存在周期长、效率低、部分性状难以测量等缺点。随着遗传学及分子生物学技术的发展,分子标记辅助选择(marker-assisted selection,MAS)应运而生;育种工作者可以直接通过检测分子标记,利用标记与目标性状基因紧密连锁的特点,选择优质基因(型)或者强性状优势个体[1]。MAS育种方法主要依赖于QTL定位的准确性,然而现有的QTL分析在寻找基因的广度、深度和精度等方面尚有不足:一是基于双亲的作图群体所含优良目标基因有限[2],二是虽然对少量效应大的遗传变异具有较好的适应性,但大部分重要的性状还受大量微效基因的协同作用,且容易受环境的影响[3]。近年来,由于高通量测序技术的迅猛发展,开发了大量丰富且廉价的SNP标记,全基因组关联分析(Genome-wide association study,GWAS)成为一种新型的MAS方法[4-5]。与QTL分析相比,GWAS通过对全基因组进行扫描,鉴定出与目标性状有关联的标记,不需要构建专门的作图群体,能同时检测多个序列变异,实现QTL的精细定位[6]。但由于受群体结构、因避免假阳性而采用严格显著性阈值等因素的影响,只有少数显著位点被利用。针对数量性状由微效多基因控制这一育种问题,基因组选择方法利用覆盖全基因组的分子标记,对个体进行了遗传评估与选择。相对于GWAS,GS方法充分反映了目标性状的遗传变异,不需要做显著性检验,即使标记效应微小,其导致的遗传变异也能被捕获。2001年Meuwissen等利用模拟数据通过GS方法对育种值的估计准确性达到85%[7]。本文对GS的原理和方法进行归纳,并基于影响GS的各种因素探讨了提高选择准确性的各种途径,展望了GS在作物育种的应用前景,旨在为GS在作物育种中的应用提供一定的参考。
1 基因组选择的原理和基本策略
基因组选择的概念最早由Haley和Visscher在第六届世界遗传学应用于畜牧生产国际会议上提出[8],并最早于2001年由Meuwissen等完善并应用于动物育种模拟数据分析中[7]。GS根据连锁不平衡原理(linkage disequilibrium,LD),假设影响性状的每个基因至少与一个标记紧密连锁,从而用该标记间接反映基因的效应[9]。GS的实现主要分3个步骤:首先,对参与分析的所有个体,利用覆盖其全基因组范围的分子标记进行基因型分型、过滤,获得基因型数据;其次,选择合适的训练群体(Training Population,TP),调查其表型,根据表型和基因型数据构建数学模型,估计每个分子标记的效应;再次,利用这些效应值对仅有基因型数据的育种群体(Breeding Population,BP)估计其对应的基因组育种值(Genomic Estimated Breeding Value,GEBV),并筛选出育种值较大的个体。基因组选择考虑了全基因组范围的SNP标记,有效地提高了选择的准确性,尤其是对低遗传力的数量性状[10]。基因组选择不需要测量所有个体的表型,可以同时对多个性状进行选择,显著地提高了育种效率,降低成本。
2 育种值估计方法
基因组选择的核心是育种值的估计,目前用于GS的方法主要分3类。
2.1 基于混合线性模型的BLUP(Best Linear Unbiased Prediction)方法
BLUP主要是通过系谱构建个体亲缘关系矩阵A,然后基于表型和系谱A,利用混合线性模型(Mixed Linear Model,MLM)计算个体的估计育种值(Estimated Breeding Value,EBV)。目前基于BLUP的GS方法主要有两种:一种是通过已测定的基因型计算个体间的相关关系矩阵G,然后用G代替系谱关系矩阵A,估算个体的育种值,即GBLUP(Genomic Best Linear Unbiased Prediction)方法[11];另一种是基于等位基因效应的RRBLUP(Ridge Regression Best Linear Unbiased Prediction),该法将标记效应作为随机效应,假定标记效应服从标准正态分布,有共同的方差,然后利用混合模型求解,每个个体的育种值等于其所在基因组标记效应的总和[7]。全基因组选择中因变量数p(标记数)往往大于反应变量数n(个体数),这种情况下常规的最小二乘估计法预测能力较低,很可能导致多重共线性和过度拟合[12],岭回归就是一种专用于共线性数据分析的改良最小二乘法,用于提高预测的准确性。当个体效应矩阵等于标记系数矩阵与标记效应乘积时,GBLUP与RRBLUP等同。
2.2 基于MCMC(Markov chain Monte Carlo)和Gibbs抽样的贝叶斯(Bayes)方法
在全基因组SNP中,并不是所有标记方差都相同。另外,只有少数SNP与影响性状的QTL连锁,其效应值或大或小;多数SNP与QTL不连锁,没有效应[13]。基于此,研究者提出了Bayes方法[7]。BayesA假设每个SNP都有特定的方差,且方差服从逆卡方分布;BayesB对标记进行了选择,认为大部分位点(π)没有遗传方差,而少数标记(1-π)有各自不同的效应方差,方差服从逆卡方分布[7];BayesC将π作为未知参数,假定其服从均匀分布U(0,1);BayesCπ在BayesC的基础上,假定标记效应方差相同;BayesDπ在BayesC的基础上,将逆卡方分布中的尺度参数作为未知参数[14];贝叶斯压缩法(Bayesian least absolute shrinkage and selection operator,Bayesian LASSO)相对于BayesB来说,假定标记效应服从双指数分布[15]。根据以上的描述发现,贝叶斯方法的改进主要是在经典BayesA和BayesB的基础上,或优化参数,或变更标记效应先验分布。贝叶斯方法在某些性状上优于BLUP方法[7,16],但仍存在计算时间长、先验分布超参数优化等问题。
2.3 人工智能领域的机器学习方法
目前常用的GS机器学习方法主要有支持向量机(support vector machine,SVM)、随机森林(Random Forest,RF)、人工神经网络(Artificial Neural Network,ANN)等[17-18]。机器学习不需要假设标记效应的分布和方差,依靠训练和学习过程,挖掘事物内部存在的规律,考虑了标记间的线性及非线性关系,在某些性状上优于传统参数方法[17,19]。尽管如此,GS方法预测效果依然无法令人完全满意,仍需根据不同的情况进行改进,并依据不同的标记、不同的性状等选择不同的方法。
3 GS的影响因素
影响GS预测准确性的主要因素有统计方法、标记密度和类型、性状遗传力的大小、训练群体的大小、训练群体和育种群体之间的关系、连锁不平衡程度等。其基本公式表述如下[20]:
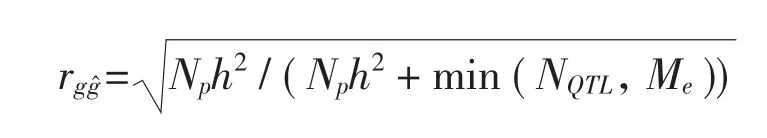
3.1 标记的选择
GS的原理是利用与基因或QTL紧密连锁的标记,用标记效应反映基因效应,因而筛选标记的方法主要有:根据LD值过滤冗余SNP或者选择tag SNP或者block tag SNP,利用GWAS筛选与性状显著相关的significant SNP。另外,标记密度越高,预测的准确性越大;标记的密度主要决定于LD的跨度及样本大小,异花授粉植物的LD跨度低于自花授粉,需要更高密度的分子标记,标记密度玉米>高粱>水稻[21]。当标记达到一定数目时,预测准确性处于平衡状态,而后增加或减少标记数目,可能会降低准确性[22]。标记的类型对GS预测的准确性有很大的影响。单倍型是由数个相邻SNP组成,更能捕获QTL,优于SNP标记模型,但是当LD程度足够大时,两者之间没差别[23]。但是SNP等位基因具有二态性,其结果的重复性较好,且使用起来更方便。
3.2 训练群体
对于训练群体的选择主要考虑TP的遗传结构、TP的大小、TP与BP的亲缘关系远近。TP的遗传多样性越丰富,预测准确性越高[24]。由于自交繁殖或者双亲杂交产生后代等因素,植物种群的遗传基础较动物狭窄,动物需要的训练群体大于植物;自然群体样本大于双亲群体。群体结构是影响GS预测准确性的一个重要因素。Isidro等[25]和Giovanny[26]阐述了基因组选择过程中如何在考虑群体结构的情况下选择最优训练集,包括随机抽样法、基于PCA的相似法、最大决定系数平均值法、最小预测误差方差平均值法、混合法等。结果表明,不同的作物、不同的性状、不同的群体结构,最优训练集的选择标准不一样。另外,TP和BP相关越强,预测的准确性越高。研究表明,对于多亲本的小麦单交群体,以下群体预测的准确性从高到低依次为TP与BP来源于共同父母本、TP与BP共享双亲之一、TP与BP没有共同双亲[27]。
3.3 统计方法
目前常用于GS预测的模型其准确性取决于个体间相关关系、分子标记效应及方差的假设分布。不同的群体、不同的性状、不同的模型,其准确性不同。模拟数据研究表明,准确性BayesB>BayesA>RRBLUP[7]。在抗小麦锈病育种中,Bayesian Lasso和RRBLUP比SVM模型表现更好[28]。对于BLUP法,考虑到混合线性模型对具有较大效应的标记效应收缩得厉害,可以将此类标记作为固定效应,在一定程度上可以提高预测准确性。研究发现,将GWAS分析筛选出来的显著与性状相关的标记作为固定效应,放入GS模型中可以提高GEBV的准确性,尤其是GBLUP法[26,29-30]。另外,结合GWAS结果提供的信息构建标记权重对角矩阵,不同的SNP赋予不同的权重,并将所有标记分为两组,一组位于已关联到的QTL区域标记,一组为未关联到的标记)也可以提高GEBV的准确性[31]。GS估计育种值往往只考虑加性效应,但是标记间可能存在一定的相关性,存在上位效应,并且对于F1群体,还需要考虑到显性效应。当非加性遗传方差存在时,在模型中如不加以考虑,将会高估加性效应。研究表明,当显性方差占表型总方差的比例达到0.2,包含加性和显性效应的预测模型显著优于加性模型[32]。GBLUP和传统的BLUP法分别从基因组和系谱的角度计算了个体间的相关关系,对于既有系谱关系又有基因组数据的群体,可以考虑将系谱和基因组数据相结合,完善关系矩阵,提高GEBV的准确性。从性状表型的角度考虑,对于那些不符合正态分布或者离散型性状,我们还可以考虑广义混合线性模型用于提高预测的准确性。
4 基因组选择在作物育种中的应用
自基因组选择的概念提出以来,GS方法在动物育种中应用越来越广泛,如小鼠、牛、鸡等,尤其是奶牛,在全球各国的奶牛产业中均得到应用[33-36]。近年来,GS在植物育种中也逐渐引起了重视,已相继开展了多种作物的GS模拟和验证实验,2007年Piyasatian等[37]用自交系杂交模拟了选择效率,结果表明GS效率高于传统的MAS。GS在玉米育种中使用最为广泛,Riedelsheimer等[38]、Zhao等[39]、Guo[40]等在玉米自交、测交、杂交F1、RIL等群体中,使用RRBLUP模型,针对玉米籽粒含水量、产量、一般配合力(General Combining Ability,GCA)、代谢物含量等性状,对个体GEBV进行了估计。GS在小麦育种中也有较多的涉猎,如Heffner等[22]的研究表明,在小麦双亲育种群体中,随着高通量基因型分型技术的发展,GS较表型选择和传统MAS节约成本、缩短育种年限;Crossa等[41]对599个小麦品系产量进行GS分析,结果表明GS比传统的BLUP预测能力提高了7.7%~35.7%;Zhao等[42]利用RRBLUP、BayesA、BayesB、BayesC以及BayesCπ方法,考虑加性和显性效应,对90个小麦杂交种进行了GS分析。GS在水稻、大麦、甘蔗等其他作物中也有研究报道[43-45]。
基因组选择方法的主要目的是对个体育种值进行准确的估计。针对不同的群体,GS还可以用于对所有遗传变异和遗传效应进行检测和估计[46],以及杂种优势的预测[47]。TP与BP亲缘关系越近,GS预测准确性越高。对与TP间隔世代的BP进行预测,GS仍具有一定的预测力。研究表明,在与BP间隔3代以内,GS准确率每世代下降约5%[7],3代以后再重新估计标记效应。利用GS还可以对个体的适应性及稳产性进行估计。农业生产中对品种的评价,不仅需要考虑高产性,还需要考虑适应性及稳产型。GS在作物育种中的应用前景广阔,但其发展仍处于初级阶段,面临着众多挑战,如不同的群体需要选择不同的模型,基于基因与环境的互作、群体结构等因素需要改善模型等,这些都需要进一步完善。基于以上总结和探索,可以考虑构建一个GS平台,一方面加强与各单位的合作,收集的丰富的表型和基因型数据;另一方面,可以收集已有的QTL信息,改善模型,将其融入GS分析当中。GS平台的目的不仅用于育种值的估计,还可用作遗传变异和效应估计、杂种优势预测、品种评价。
[1]Xu Y B,Crouch J H.Marker-assisted selection in plant breeding:from publications to practice[J].Crop Science,2008,48(2):391-407.
[2]孔繁玲.植物数量遗传学[M].北京:中国农业大学出版社,2006.
[3]Rex B.Molecular markers and selection for complex traits in plants:learning from the last 20 years[J].Crop Science,2008,48(5):1649-1664.
[4]Atwell S,Huang Y S,Vilhjalmsson B J,et al.Genome-wide association study of 107 phenotypes in a common set of Arabidopsis thaliana inbred lines[J].Nature,2010,465(7298):627-631.
[5]Huang X H,Zhao Y,Wei X H,et al.Genomewide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm[J].Nature Genetics,2012,44(1):32-39.
[6]Zhu C S,Michael G,Yu J M,et al.Status and prospects of association mapping in plants[J].The Plant Genome,2008,1(1):5-20.
[7]Meuwissen T H E,Hayes B J,Goddard M E.Prediction of total genetic value using genomewide dense marker maps[J].Genetics,2001,157(4):1819-1829.
[8]Meuwissen T H E.Genomic selection:marker assisted selection on a genome wide scale[J].Journal of Animal Breeding and Genetics,2007,124(6):321-322.
[9]Goddard M E,Hayes B J.Mapping genes for complex traits in domestic animals and their use in breeding programmes[J].Nature Reviews Genetics,2009,10(6):381-391.
[10]Resende R M S,Casler M D,de Resende M D V.Genomic selection in forage breeding:accuracy and methods[J].Crop Science,2014,54(1):143-156.
[11]VanRaden P M.Efficient methods to compute genomic predictions[J].Journal of Dairy Science,2008,91(11):4414-4423.
[12]唐金梅,陈建国.全基因组选择在植物育种中的研究进展[J].贵州农业科学,2016,44(8):1-5.
[13]王重龙,丁向东,刘剑锋,等.基因组育种值估计的贝叶斯方法[J].Hereditas(Beijing),2014,36(2):111-118.
[14]Habier D,Fernando R L,Kizilkaya K,et al.Extension of the bayesian alphabet for genomic selection[J].BMC Bioinformatics,2011,12(1):1-12.
[15]Yi N J,Xu S Z.Bayesian LASSO for quantitative trait loci mapping[J].Genetics,2008,179(2):1045-1055.
[16]Moser G,Tier B,Crump R E,et al.A comparison of five methods to predict genomic breeding values of dairy bulls from genome-wide SNP markers[J].Genetics Selection Evolution,2009,41(1):1-16.
[17]Ornella L,Perez P,Tapia E,et al.Genomicenabled prediction with classification algorithms[J].Heredity,2014,112(6):616-626.
[18]Ogutu J O,Piepho H P,Schulz-Streeck T.A comparison of random forests,boosting and support vector machines for genomic selection[J].BMC Proceedings,2011,5(3):1-5.
[19]束永俊,吴磊,王丹,等.人工神经网络在作物基因组选择中的应用[J].作物学报,2011,37(12):2179-2186.
[20]Daetwyler H D,Pongwong R,Villanueva B,et al.The impact of genetic architecture on genomewide evaluation methods[J].Genetics,2010,185(3):1021-1031.
[21]Gupta P K,Rustgi S,Kulwal P L.Linkage disequilibrium and association studies in higher plants:present status and future prospects[J].Plant Molecular Biology,2005,57(4):461-485.
[22]Heffner E L,Jannink J L,Iwata H,et al.Genomic selection accuracy for grain quality traits in biparental wheat populations[J].Crop Science,2011,51:2597-2606.
[23]Calus M P L,Veerkamp R F.Accuracy of breeding values when using and ignoring the polygenic effect in genomic breeding value estimation with a marker density of one SNP per cM[J].Journal of Animal Breeding and Genetics,2007,124(6):362-368.
[24]Heslot N,Yang H P,Sorrells M E,et al.Genomic selection in plant breeding:a comparison of models[J].Crop Science,2012,52:146-160.
[25]Isidro J,Jannink J L,Akdemir D,et al.Training set optimization under population structure in genomic selection[J].Theoretical and Applied Genetics,2015,128(1):145-158.
[26]Giovanny C P.Genome-assisted prediction of quantitative traits using the R package sommer[J].Plos One,2016,11(6):1-15.
[27]Technow F,Schrag T A,Schipprack W,et al.Genome properties and prospects of genomic prediction of hybrid performance in a breeding program of maize[J].Genetics,2014,197(4):1343-1355.
[28]Ornella L,Sukhwinder S,Perez P,et al.Genomic prediction of genetic values for resistance to wheat rusts[J].The Plant Genome,2012,5(3):136-148.
[29]Rex B.Genomewide selection when major genes are known[J].Crop Science,2014,54(1):68-75.
[30]Abdollahi-Arpanahi R,Morota G,Valente B D,et al.Assessment of bagging GBLUP for wholegenome prediction of broiler chicken traits[J].Journal of Animal Breeding and Genetics,2015,132(3):218-228.
[31]Zhang Z,Ober U,Erbe M,et al.Improving the accuracy of whole genome prediction for complex traits using the results of genome wide association studies[J].Plos One,2014,9(3):1-12.
[32]de Almeida Filho J E,Guimaraes J F R,e Silva F F,et al.The contribution of dominance to phenotype prediction in a pine breeding and simulated population[J].Heredity,2016,117(1):33-41.
[33]Legarra A,Robert-Granie C,Manfredi E,et al.Performance of genomic selection in mice [J].Genetics,2008,180(1):611-618.
[34]Hayes B J,Bowman P J,Goddard M E,et al.Invited review:genomic selection in dairy cattle:progress and challenges[J].Journal of Dairy Science,2009,92(2):433-443.
[35]Chen C Y,Misztal I,Aguilar I,et al.Genomewide marker-assisted selection combining all pedigree phenotypic information with genotypic data in one step:an example using broiler chickens[J].Journal of Animal Science,2011,89(1):23-28.
[36]Loberg A,Durr J W.Interbull survey on the use of genomic information[J].Interbull Bull,2009,39:3-14.
[37]Piyasatian N,Fernando R L,Dekkers J C M.Genomic selection for marker-assisted improvement in line crosses[J].Theoretical and Applied Genetics,2007,115(5):665-674.
[38]Riedelsheimer C,Czedik-EysenbergA,Grieder C,et al.Genomic and metabolic prediction of complex heterotic traits in hybrid maize[J].Nature Genetics,2012,44(2):217-220.
[39]Zhao Y S,Gowda M,Liu W X,et al.Accuracy of genomic selection in European maize elite breeding populations[J].Theoretical and Applied Genetics,2012,124(4):769-776.
[40]Guo T T,Li H H,Yan J B,et al.Performance prediction of F1 hybrids between recombinant inbred lines[J].Theoretical and Applied Genetics,2013,126(1):189-201.
[41]Crossa J,de losCampos G,Perez P,et al.Prediction of genetic values of quantitative traits in plant breeding using pedigree and molecular markers[J].Genetics,2010,186(2):713-724.[42]Zhao Y S,Zeng J,Fernando R L,et al.Genomic prediction of hybrid wheat performance[J].Crop Science,2013,53(3):802-810.
[43]Xu S Z,Zhu D,Zhang Q F.Predicting hybrid performance in rice using genomic best linear unbiased prediction[J].PNAS,2014,111(34):12456-12461.
[44]Zhong S Q,Dekkers J C M,Fernando R L,et al.Factors affecting accuracy from genomic selection in populations derived from multiple inbred lines:a barley case study[J].Genetics,2009,182(1):355-364.
[45]Gouy M,Rousselle Y,Bastianelli D,et al.Experimental assessment of the accuracy of genomic selection in sugarcane[J].Theoretical and Applied Genetics,2013,126(10):2575-2586.
[46]Vitezica Z G,Varona L,Legarra A.On the additive and dominant variance and covariance of individuals within the genomic selection scope[J].Genetics,2013,195(4):1223-1230.
[47]Zhao Y S,Li Z,Liu G Z,et al.Genome-based establishment of a high-yielding heterotic pattern for hybrid wheat breeding[J].PNAS,2015,112(51):15624-15629.
猜你喜欢
今日农业(2021年11期)2021-08-13
建材发展导向(2021年10期)2021-07-16
中学生数理化(高中版.高考数学)(2021年3期)2021-06-09
新世纪智能(语文备考)(2021年11期)2021-03-08
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
中学生数理化·七年级数学人教版(2019年6期)2019-06-25
中学生数理化·七年级数学人教版(2019年6期)2019-06-25
中成药(2018年7期)2018-08-04
初中生世界·九年级(2017年10期)2017-11-08
