
不同π链的芳胺类敏化剂性质的理论研究
2018-05-30 09:28郭惠霞席晓华闫任翔卢小泉
西北师范大学学报(自然科学版) 2018年2期
郭惠霞,席晓华,闫任翔,卢小泉
(甘肃省生物电化学与环境分析重点实验室,西北师范大学 化学化工学院,甘肃 兰州 730070)
如何经济有效地利用太阳能是太阳能电池研究与开发的至关重要的课题.染料敏化太阳能电池以其无污染、低耗费等优势吸引了科学家们的注意,且有望代替传统的无机太阳能电池[1].染料敏化太阳能电池使用吸附在半导体(一般采用TiO2)上的敏化剂分子来捕获太阳光,并且将电荷传输给半导体[2-3].因此,敏化剂的性质是影响染料敏化太阳能电池效率的关键因素之一.敏化剂一般分为钌配合物染料[4]、纯有机染料[5]和卟啉类敏化剂三大类[6],由于钌是贵金属且对环境污染严重,因此非金属有机敏化剂以其独特的优势得到了科学界重视.许多研究表明,通过修改具有D-π-A(D代表敏化剂供体,π通常指π共轭链,而A指敏化剂的受体)型敏化剂中π链的结构,可以改变敏化剂的吸收光谱、摩尔消光系数、光捕获效率、带隙等性质[7-8],进而改变太阳能电池的光电转化效率.
基于以上这些研究以及Scrascia等合成的AS系列敏化剂[9],文中设计了2个系列的新型敏化剂,在A系列中分别用吡咯、呋喃和苯环基团代替有机分子AS5敏化剂中的噻吩单元得到敏化剂A2、A3和A4,研究π链上不同杂环对染料敏化太阳能电池性能的影响;在B系列中,通过调换敏化剂AS5结构中π链上噻吩和苯环的相对位置得到敏化剂B1,进而检测氟原子位置对敏化剂性能的影响.在敏化剂中,氟原子在π链上的取代可有效提高太阳能电池的效率,这是由于额外的拉电子基有利于电荷的分离和供体和受体之间π链的延伸.同理,在敏化剂B1结构基础上,用噻吩、吡咯和呋喃单元替代苯环基团得到敏化剂B4、B2和B3.运用Gaussion 09程序软件包对上述敏化剂进行计算,对比所设计敏化剂与已知AS5敏化剂的几何结构、电子分布以及光物理性能等,在理论上寻找更高效的敏化剂,为实验的提供理论支撑.
1 计算方法
所有敏化剂基态几何结构运用密度泛函理论,在B3LYP/6-31G(d,p)水平下,气相中进行优化.同时在相同的理论水平上进行频率计算,结果显示优化后的几何构型处于势能面上的最小值点(没有虚频).为了更准确模拟敏化剂吸收光谱,以染料AS5为例,选取BHandHLYP、M06-HF、CAM-B3LYP、ωB97XD等方法进行测试,结果见表1,发现在杂化泛函中HF成分占比越高所计算出的激发能也越高,其中,使用ωB97XD得到的最大吸收光谱峰位置与实验值(419 nm)接近,因此,在后面的计算中采用ωB97XD结合IEFPCM模型模拟CHCl3的溶剂化效应.所有计算均在Gaussion 09程序包内完成.同时,利用Multiwfn[10]程序模拟了敏化剂紫外-可见吸收光谱及相应跃迁[11].

表1 不同方法在6-31G(d,p)水平上计算敏化剂 AS5的最大吸收波长
2 结果与讨论
2.1 π链的修改对几何结构的影响
AS5敏化剂的2个异构体在X-Functional/6-31G(d,p)(X=B3LYP,CAM-B3LYP,ωB97XD,M06-HF)水平上气态中进行基态几何优化,所得的结果见表2,从表中可以得出,E构型比Z构型稳定.这与相关文献结论一致[12],故在后面分子结构中,敏化剂均采用E构型.

表2 使用不同的方法在6-31G(d,p)水平下 计算的基态敏化剂AS5能量(eV)
图1标出了DFT-B3LYP/6-31G(d,p)理论水平上优化的相关键长及相应的二面角.所研究的敏化剂π链键长基本相近,即b1=0.142 nm,b2=0.122 nm.从图中明显看出,敏化剂B2,B3和B4的键长b3,b4,b5与其他敏化剂对应的键长相比最小.此外,敏化剂B2,B3和B4中两杂环之间的二面角相比之下也较小.这说明当噻吩基团处于靠近受体的位置时,内部电荷转移得到了提高且染料敏化太阳能电池的转化效率也得到相应提升,这是由于引入的噻吩单元靠近受体从而增加了体系的共平面性,因此电荷转移能降低[12].而在敏化剂AS5,A2,A3,A4,B1和B2中两杂环之间的二面角在11°~33°之间变动.
2.3 π链对敏化剂前线分子轨道的影响
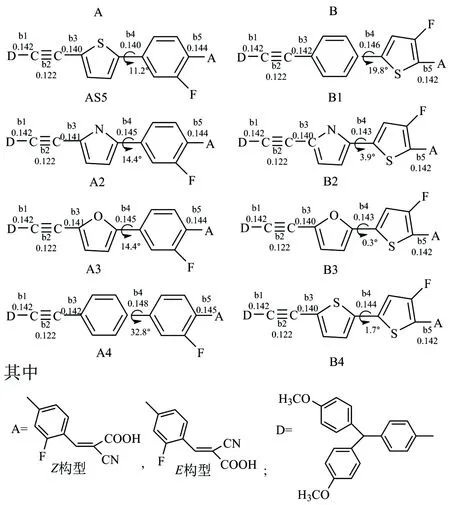
图1 分子结构框架及优化的键长(nm)键角(°)
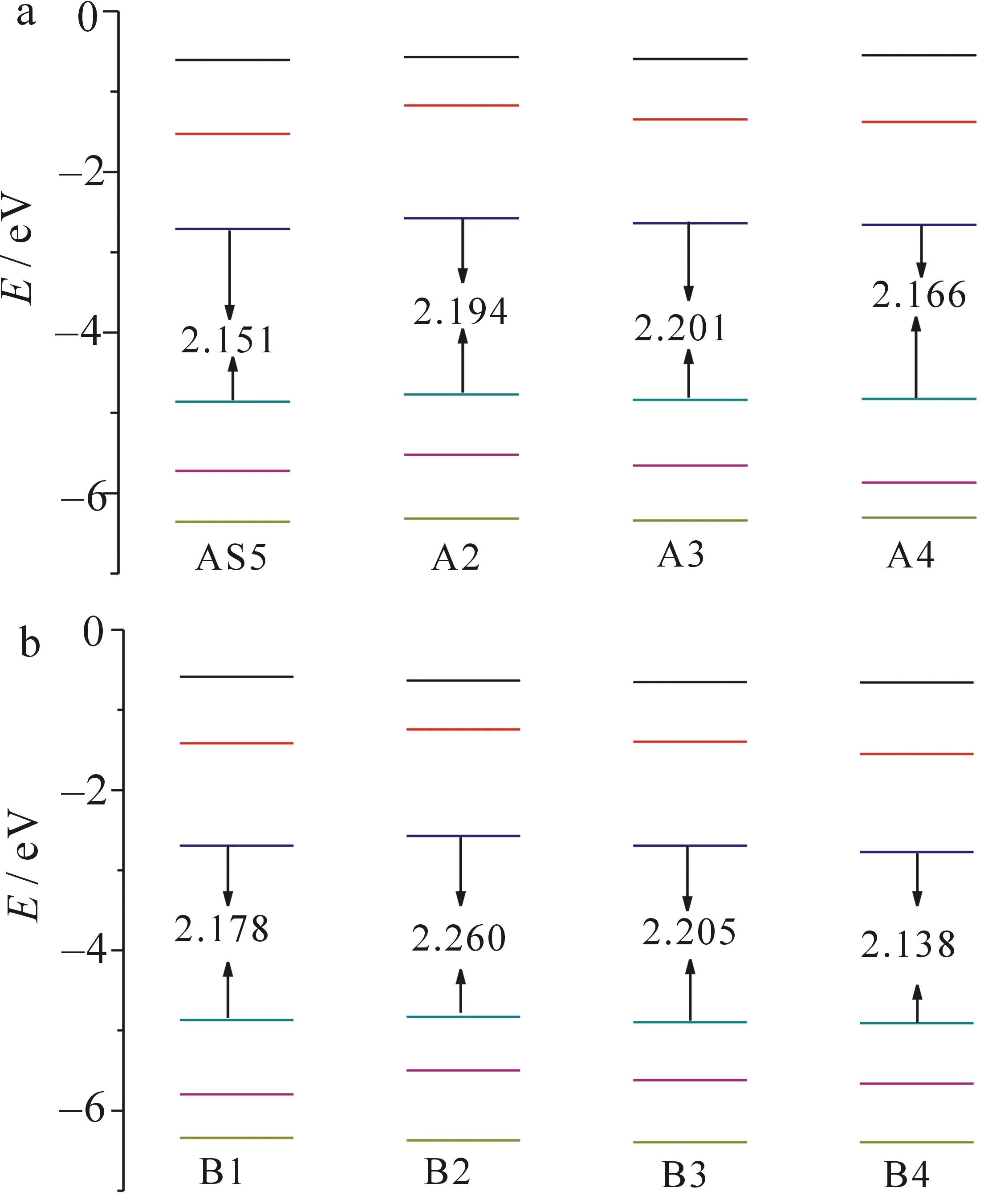
图2 敏化剂A组(a)和B组(b)的前线分子轨道能级图
图2是在B3LYP/6-31G(d,p)水平上计算的敏化剂的前线分子轨道能级图.一个高效的敏化剂应该具有电子注射快、敏化剂可以再生及窄的带隙等特点.因此,HOMO能级应该低于氧化还原电对能级, 氧化态的敏化剂才能被还原, 而LUMO能级应该高于半导体导带,处于激发态的电子才能够有效注入二氧化钛导带.从图3可以看出,所有HOMO能级值都小于电解质氧化还原电位(-4.6 eV),而所有LUMO能级值都高于二氧化钛导带值(-4.0 eV).在A系列中,吡咯、呋喃和苯环基团对噻吩的取代使LUMO值增加了约0.1 eV,而对HOMO能级值几乎没影响.除吡咯(增加了约0.1 eV)外,其他敏化剂带隙差都为0.05 eV.换而言之,杂环的替代对敏化剂带隙几乎无影响.对比敏化剂AS5与敏化剂B1,可以看出,HOMO能级值、LUMO能级值或者带隙值几乎保持不变,即π链上苯环与噻吩单元位置的替换,对前线分子轨道几乎无影响.带隙大小按照A3(2.201 eV)>A2(2.194 eV)>A4(2.166 eV)>AS5(2.151 eV)顺序排列.在B系列中,发现了与A系列类似的规律,替代杂环对敏化剂的HOMO能级无影响,同时对敏化剂B3的 LUMO能级无影响,但是吡咯环的替代使敏化剂B2的LUMO能级值增加了0.12 eV.
在所有敏化剂中,敏化剂B4带隙最小,这是由于存在双噻吩单元导致的.关于噻吩单元数量对敏化剂的影响在相关文献中也有叙述,即增加噻吩单元能够降低带隙[13].B系列带隙大小顺序为B2(2.26 eV)>B3(2.205 eV)>B1(2.178 eV)>B4(2.138 eV).表明拥有较窄带隙的敏化剂其吸收带变宽且发生红移,进而增加其光电转化效率.
此外,利用Multiwfn 3.4在ωB97XD/6-31G(d,p)水平下计算了敏化剂从基态到第一激发态的电荷密度差图,其相应的电荷转移量qCT和电荷转移距离DCT见图3.结果表明,电子密度减少的区域主要集中在供体及π链,而电子密度增加的区域分布在受体上甚至延伸到了π链,说明从基态到激发态时电荷发生了很明显的内部转移.同样的结论从电荷转移量(0.572 e~0.686 e)和电荷转移距离(0.407~0.531 nm)也可得到.
2.3 改变π链对吸收光谱的影响
高效的敏化剂应具备宽的吸收带及强的振子强度等特点.基于优化后得到的基态几何结构,通过TDDFT计算30个最低singlet singlet激发态,模拟敏化剂在三氯甲烷中的吸收光谱见图4,相应的激发态能量、振子强度、主要轨道跃迁贡献详见表3.

图3 敏化剂的电荷密度差图(紫色代表电子密度的增加,蓝色代表电子密度的减少,qCT(e)代表电荷转移量, DCT(nm)代表电荷转移距离)
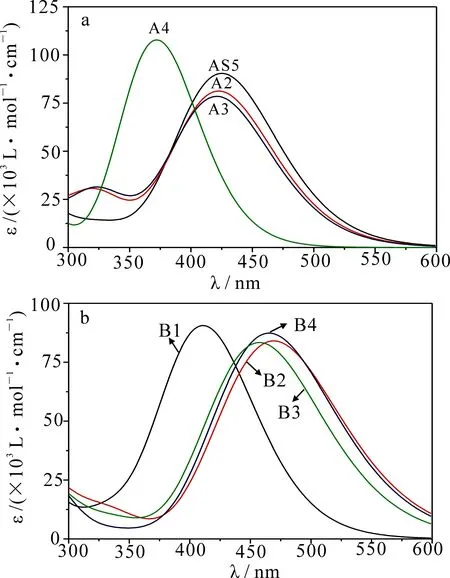
图4 敏化剂A组(a)和B组(b)的紫外可见吸收光谱图
在图4中,相比于A系列敏化剂的最大吸收波长来说,B系列除了敏化剂B1,其余敏化剂均发生了明显红移.在A系列敏化剂中,其π链中噻吩单元用吡咯单元和呋喃单元替换后并没有改变敏化剂的最大吸收波长,而采用苯基替代则明显发生了蓝移(敏化剂A4的最大吸收波长为372 nm,而敏化剂AS5、A2和A3的最大吸收波长分别为425,423和421 nm).π链上杂环种类与敏化剂最大吸收波长的关系在其他文献中也有类似的结论,将双噻吩和两个杂环作为π链时,其相应敏化剂的最大吸收波长红移最大[14].敏化剂最大吸收波长的顺序为AS5(425 nm)>A2(422 nm)>A3(421 nm)>A4(372 nm),与相应敏化剂带隙相比其顺序不一致,这主要是因为敏化剂从基态向第一激发态跃迁时主要贡献不仅仅只是由HOMO到LUMO的跃迁,而且还有HOMO-1到LUMO的跃迁.在B系列中,由于将噻吩单元与苯环单元的顺序进行调换,使敏化剂B1的最大吸收波长比敏化剂AS5明显蓝移了14 nm,而将噻吩单元、吡咯单元和呋喃单元对苯基进行替换,则会导致敏化剂B2,B3,B4的最大吸收波长发生明显红移,换言之,只有用氟进行单取代且双杂环作为π链时才会明显提高敏化剂的最大吸收波长,进而提高光捕获效率(ELH).B系列的最大吸收波长顺序依次为B2(469 nm)>B4(465 nm)>B3(457 nm)>B1(411 nm).从A系列和B系列发现,有噻吩基团的敏化剂比无噻吩基团的敏化剂吸收带宽,这是由于硫原子的存在使其成为富电子体系,而且具有良好的平面规整性和比较高的电子迁移率.此外,增加噻吩单元的长度,能够改善分子的结晶性,进而增加分子间的堆积效应,使得分子的吸收谱带变宽,最终达到提升DSSCs的光电转化效率.从AS5和B4,A2和B2,A3和B3,A4和B1的吸收光谱比较发现,敏化剂中氟原子对噻吩单元的取代更有利于敏化剂吸收谱带的拓宽.

表3 敏化剂相关参数
2.5 敏化剂光电转化效率
染料敏化太阳能电池能量转化效率η与短路电流密度JSC、开路电压VOC、填充因子(FF),以及太阳能强度(Pinc)有密切联系,其关系可表示为:
(1)
其中短路电流密度又可表示为
(2)
其中,ELH(λ)为特定波长的光捕获效率;Φinj为电子注射效率;ηcoll为电荷收集效率;ηreg为染料再生效率.从(1)和(2)式,可以得出ELH,其中ηcoll和ηreg越大,JSC越大,η就越大.ELH可以由如下公式得出
ELH(λ)=1-10-f,
(3)
f为确定波长处的振子强度,即f越大,ELH越大.从表1的数据可以明显看出所有敏化剂都具有较高的振子强度,也就意味着敏化剂在最大吸收波长处的光捕获效率值都应该较高,计算后发现其值达到了99%.由(2)式可知,吸收带宽度越宽,可导致敏化剂的JSC增大,故敏化剂B2,B3和B4的JSC比较高.由于敏化剂的供体受体相同,在相同的实验条件下,假设电子注射效率、电荷收集效率和染料再生效率为常数,那么预测敏化剂B2、B3和B4的JSC将比敏化剂AS5的高,而实验测得到AS5的JSC为10.65 mA·cm-2,所以B2,B3和B4的JSC将超过10.65 mA·cm-2.
基于Marcus理论,电子注射率Φinj可以通过电子注射自由能(电子注射驱动力)来衡量.注射驱动力越大越有利于电子注射,进而提高太阳能电池工作效率.从激发态到TiO2的电子注射驱动力可以用下列公式表示:
ΔGinj=Edye*-ECB=Edye-λmax-ECB,
(4)
其中,Edye*为激发态敏化剂的氧化势能;ECB为TiO2的还原电位(采用4.0 eV);Edye为基态敏化剂的氧化电势(其大小约等于-EHOMO),而λmax为垂直激发能.当ΔGinj为负数时,电子可以自发地从敏化剂注射到TiO2.
敏化剂再生效率ηreg一般通过敏化剂的再生驱动力进行评估,其公式如下:
ΔGreg=Eredox-Edye,
(5)
其中,ΔGinj为敏化剂的再生驱动力;Eredox大小一般为4.6 eV.
为了研究π链的改变对短路电流密度的影响,在B3LYP/6-31G(d,p)水平下优化几何结构的基础上,进一步采用TD-ωB97XD/6-31G(d,p)计算了电子注射驱动力(ΔGinj)及敏化剂再生驱动力(ΔGreg),结果见表1.敏化剂的ΔGinj在 1.754~2.163 eV范围内,其值为负值表明敏化剂激发态的电子可以自发的注入到TiO2导带中,同时ΔGreg绝对值大于0.2 eV,说明超过90%的敏化剂可以再生,电子的注射效率也超过了90%[15].综上所述,由于影响敏化剂的光电转化效率主要是吸收谱带的宽度,因此敏化剂B2、B3和B4的η有望超过敏化剂AS5的η(5.81%).
3 结论
利用DFT和TDFDT方法,对8种敏化剂的光捕获效率、分子内部电荷转移效率、电子注射效率、敏化剂再生效率等相关重要参数进行了对比研究后得出:对于AS5类敏化剂来说,锚定基团的E构型比Z构型稳定;其次,将新设计的敏化剂与敏化剂AS5相比,敏化剂B2,B3和B4的紫外可见吸收光谱延伸到460 nm附近,表现出了明显的电荷分离情况.而当苯环处于靠近锚定基团的位置时(A系列敏化剂),其对应敏化剂的吸收谱带于噻吩单元靠近锚定基团时(B系列敏化剂);再次,新设计的敏化剂光捕获效率达到了99%,电子注射效率和敏化剂再生效率达到了90%,即敏化剂B2,B3,B4有望成为高效敏化剂的备选物,为在试验中合成新的敏化剂提供了理论指导.
参考文献:
[1] NING Zhi-jun,FU Ying,TIAN He.Improvement of dye-sensitized solar cells:what we know and what we need to know[J].EnergyEnvironSci,2010,3(9):1170.
[2] TZEL M.Conversion of sunlight to electric power by nanocrystalline dye-sensitized solar cells[J].JPhotochemPhotobiolA,2004,164(1):3.
[3] LI Lu-lin,CHANG Yu-cheng,WU Hui-ping,et al.Characterisation of electron transport and charge recombination using temporally resolved and frequency-domain techniques for dye-sensitised solar cells[J].InternationalReviewsinPhysicalChemistry,2012,31(3):420.
[4] PREZLE N C,KADOR L,PENG B,et al.Characterization of the adsorption of Ru-Bpy dyes on mesoporous TiO2films with UV-Vis,Raman,and FTIR spectroscopies[J].JPhysChemB,2006,110(17):8723.
[5] TIAN H,YANG X,CHEN R,et al.Phenothiazine derivatives for efficient organic dye-sensitized solar cells[J].ChemCommun,2007,36:3741.
[6] BERR C,CARDENAS G I,MARCO J F,et al.Theoretical and spectroscopic study of nickel(II) porphyrin derivatives[J].JPhysChemA,2007,111(14):2706.
[7] CHITPAKDEE C,NAMUANGRUK S,SUTTISINTONG K,et al.Effects ofπ-linker,anchoring group and capped carbazole at meso-substituted zinc-porphyrins on conversion efficiency of DSSCs[J].DyesandPigments,2015,118:64.
[8] CHITPAKDEE C,JUNGSUTTIWONG S,SUDYOADSUK T,et al.Modulation ofπ-spacer of carbazole-carbazole based organic dyes toward high efficient dye-sensitized solar cells[J].SpectrochimActa.PartA,2017,174:7.
[9] SCRASCIA A,DE MARCO L,LARICCHIA S,et al.Fluorine-thiophene-substituted organic dyes for dye sensitized solar cells[J].JMaterChemA,2013,1(38):11909.
[10] LU Tian,CHEN Fei-wu.Multiwfn:a multifunctional wavefunction analyzer[J].JComputChem,2012,33(5):580.
[11] LU Tian,CHEN Fei-wu.Calculation of molecular orbital composition[J].ActaChimicaSinica,2011,69(20):2393.
[12] HASANEIN A A,ELMARASSI Y R,KASSEM E N.TD-DFT investigation of D-π-A organic dyes with thiophene moieties asπ-spacers for use as sensitizers in DSSCs[J].JMolecularModeling,2016,22(5):1.
[13] MEHMOOD U,HUSSEIN I A,DAUD M,et al.Theoretical study of benzene/thiophene based photosensitizers for dye sensitized solar cells(DSSCs)[J].DyesandPigments,2015,118:152.
[14] GUO Y,LU X,LI G,et al.Theoretical design of push-pull porphyrin dyes withπ-bridge modification for dye-sensitized solar cells[J].JPhotochemPhotobiolA,2017,332:232.
[15] ISLAM A,SUGIHARA H,ARAKAWA H.Molecular design of ruthenium(II) polypyridyl photosensitizers for efficient nanocrystalline TiO2solar cells[J].JPhotochemPhotobiolA,2003,158(2):131.
猜你喜欢
煤炭学报(2022年11期)2023-01-07
河北科技大学学报(2020年3期)2020-07-14
石油与天然气化工(2020年1期)2020-04-16
石油与天然气化工(2019年1期)2019-03-06
中国资源综合利用(2017年1期)2018-01-22
有色金属材料与工程(2017年2期)2017-05-31
山东工业技术(2016年15期)2016-12-01
科技资讯(2016年5期)2016-08-13
中国粮油学报(2016年5期)2016-01-23
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
