
Mo2C(101)上甲醇解离反应机理的理论研究
2019-01-24 02:59,
山东化工 2019年1期
,
(山东临沂大学 化学化工学院,山东 临沂 276000)
甲醇(CH3OH)是结构最简单的饱和一元醇,科研工作者已经在很多过渡金属表面研究了甲醇的吸附和解离:包括Ni(100),Cu(111),Ag(111),Au(111),Ru(001)等[1-3]。
近年来,碳化钼催化剂因为其优异的催化性能引起人们的广泛关注,它在很多反应中起到重要的催化作用:如制氢反应、氧化还原反应、甲醇重整反应和费托合成反应等,在这些反应中碳化钼的催化活性与贵金属相当,有的催化活性甚至会超越贵金属催化剂。
由于碳化钼催化剂在很多反应中具有良好的催化性能,所以在理论上研究甲醇在该催化剂上的解离反应机理,从而推断出最佳反应路径,这对将来的实验研究以及实际的生产生活具有重要的理论指导意义。
1 计算方法和模型
1.1 计算方法
所有计算采用VASP软件[4],利用密度泛函理论中的PBE交换相关泛函,对体系进行能量计算与结构优化。采用CI-NEB方法[5]对过渡态进行搜索。
1.2 计算模型
在本工作中,选择六方Mo2C最稳定的(101)面进行研究,其结构如图1所示。
Ea=ETS-EIS
(公式1)
Er=EFS-EIS
(公式2)
其中,EIS、ETS和EFS分别表示反应始态(IS)、过渡态(TS)和终态(FS)的能量。
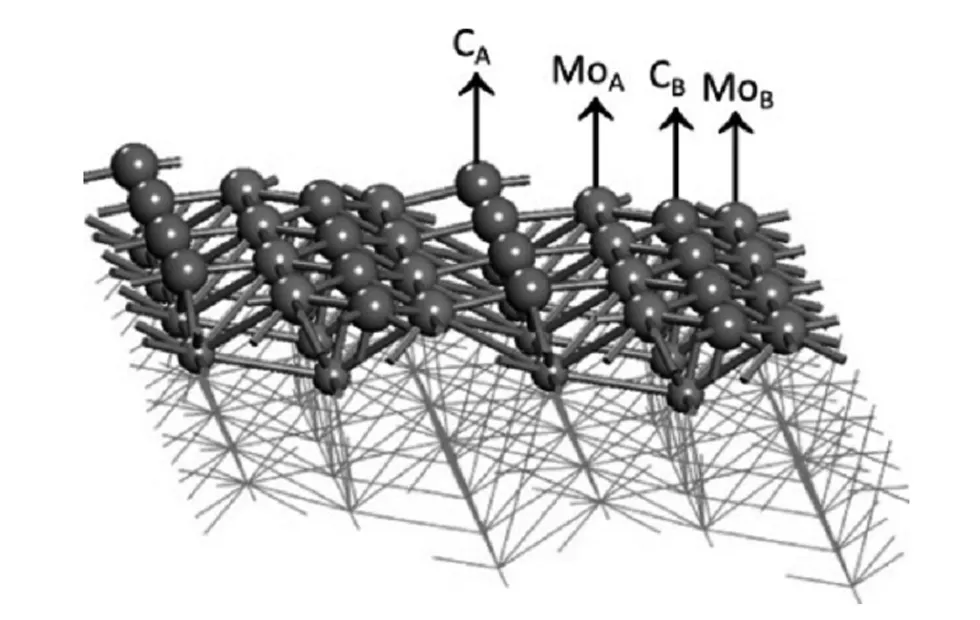
图1 Mo2C(101)表面的侧视图
2 结果与讨论
2.1 甲醇解离的反应机理
2.1.1 甲醇解离生成CH3O+H,CH2OH+H,CH3+OH
甲醇的第一步解离反应,有如下几种可能路径:(a)O-H键的断裂(R1:CH3OH→CH3O+H);(b)C-H键的断裂(R2:CH3OH→CH2OH+H);(c)C-O键的断裂(R3:CH3OH→CH3+OH)。计算得到的反应始态、过渡态、终态的结构如图2所示。

图2 R1-R3反应中优化结构的俯视图
Fig. 2 Top views of the optimized geometries for the reaction routes R1-R3

2.1.2 CH3O+H解离生成CH2O+2H和CH3+O+H

图3 R4-R5反应中优化结构的俯视图
在最有利的反应路径R1(CH3OH→CH3O+H)的基础上,以CH3O和H的共吸附作为反应的初态,考虑如下两种平行反应:(a)C-H键的断裂(R4:CH3O+H→CH2O+2H);(b)C-O键的断裂(R5:CH3O+H→CH3+O+H)。计算得到结构图如图3所示。
2.1.3 CH2O+2H解离生成CH2+O+2H和CHO+3H
以CH2O和2H为反应初态,同理考虑了如下两种情况:C-O键的断裂(R6:CH2O+2H→CH2+O+2H)和C-H键的断裂(R7:CH2O+2H→CHO+3H),图4给出了优化的结构图。
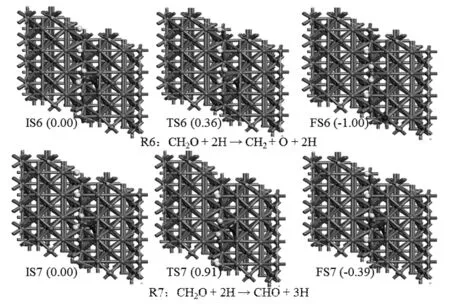
图4 R6-R7反应中优化结构的俯视图
Fig.4 Top views of the optimized geometries for the reaction routes R6-R7

2.1.4 C+O+4H的生成

图5 R8-R9反应中优化结构的俯视图

2.2 势能面
在上面讨论的基础上,绘制了整个反应的势能面如图6所示。

图6 甲醇在Mo2C(101)面上反应的势能面(能量以eV单位)
Fig.6 Potential energy surfaces (in eV) for CH3OH dissociation on the Mo2C(101) surface
综上所述,甲醇在Mo2C(101)面解离的第一步有三个平行反应,分别为:(a)O-H键之间的断裂(R1:CH3OH →CH3O+H);(b)C-H之间的断裂(R2:CH3OH→CH2OH+H);(c)C-O之间的断裂(R3:CH3OH→CH3+OH)。其中,生成CH3+OH的能垒最高(1.73 eV),生成CH2OH+H的能垒是0.96 eV,而生成CH3O+H的能垒最低,只有0.36 eV,根据能量最低原理,甲醇解离生成CH3O+H是最有可能的。生成的CH3O+H继续解离有两个平行反应:C-H键的断裂(生成CH2O+2H)和C-O键的断裂(生成CH3+O+H)反应。由于前者的反应能垒低于后者(1.23eV vs. 1.99 eV),因此前者的反应可能性更大。CH2O+2H继续解离,生成CH2+O+2H的能垒是0.36 eV,而生成CHO+3H的能垒是0.91 eV。所以在CH2+O+2H的基础上考虑后续反应。最后两个C-H键断裂的能垒分别为1.27 和1.05 eV,反应均为吸热反应。由此可见,最后两个C-H键的断裂相对较难,但是在一定外界压力和温度条件下,该反应是可以发生的。综上所述,甲醇在Mo2C(101)面上解离的最低反应路径为CH3OH→CH3O+H→CH2O+2H→CH2+O+2H→CH+O+3H→C+O+4H。
3 总结
本文采用密度泛函理论方法对甲醇在Mo2C(101)面上的反应机理进行了理论研究,计算了所有可能反应路径的活化能(Ea)以及反应热(Er)。计算结果表明,在Mo2C(101)面上,根据能量最低原理,甲醇解离的最优路径为:CH3OH→CH3O+H→CH2O+2H→CH2+O+2H→CH+O+3H→C+O+4H。这对将来的实验研究以及实际的生产生活具有重要的理论指导意义。
猜你喜欢
北京航空航天大学学报(2022年5期)2022-06-06
华东理工大学学报(自然科学版)(2022年2期)2022-04-29
中学课程辅导·教学研究(2021年8期)2021-07-14
化学工业与工程(2021年3期)2021-06-25
当代陕西(2019年17期)2019-10-08
电脑知识与技术(2018年3期)2018-03-21
哈尔滨理工大学学报(2017年1期)2017-04-08
安徽农学通报(2016年24期)2017-01-12
考试周刊(2016年88期)2016-11-24
科技视界(2016年24期)2016-10-11
