
一对戊二酸血症型双胞胎兄妹诊治
2019-04-03 01:58李艳云田凤艳
郑州大学学报(医学版) 2019年2期
甄 诚,李艳云,田凤艳
郑州大学第一附属医院儿科 郑州 450052
戊二酸血症Ⅱ型为常染色体隐性遗传病,为脂肪酸代谢紊乱性疾病,又称多种酰基辅酶A脱氢酶缺乏症(multipleacyl-CoAdehydrogenasedeficiency,MADD),较为罕见。因编码线粒体遗传性电子转运黄素蛋白或电子转运黄素蛋白脱氢酶(ETFDH)缺陷造成呼吸链多种脱氢酶功能障碍,导致脂肪酸、支链氨基酸、维生素、能量代谢障碍、中间代谢产物蓄积而引起的一系列临床症状[1]。作者对郑州大学第一附属医院儿科收治的2例误诊的戊二酸血症Ⅱ型病例资料进行分析,以便加强临床医生对戊二酸血症Ⅱ型的认识。
1 临床资料
双胞胎兄妹,11岁,均以“间断恶心、呕吐3a,加重2个月”入院。生长发育正常。神志清,精神差,淡漠面容,咽腔充血,四肢无畸形。兄:双上肢肌力5级、双下肢肌力4级;妹:四肢肌力及肌张力正常。实验室检查:血气分析pH7.15(兄)/7.20(妹),乳酸6.10(兄)/8.50(妹)mmol/L,碱剩余-12.50(兄)/-9.70(妹)mmol/L。兄血常规:白细胞5.20×109个/L、血红蛋白98.0g/L、网织红细胞4.37%、血小板总数450×109个/L;妹血常规正常。尿常规:酮体()(兄)/()(妹),尿比重1.005(兄)/1.020(妹)。空腹血糖3.34mmol/L(兄)/5.40mmol/L(妹)。同型半胱氨酸、叶酸、维生素B12、血氨、电解质、肝肾功、心肌酶、血脂、铜蓝蛋白、铁三项、病毒全套、C反应蛋白、血小板、粪常规等无异常。兄腹部超声示肝右叶钙化灶、右叶内可见一强回声光斑(直径约6mm×4mm),妹正常。兄、妹心脏超声、心电图均提示正常。头颅MRI示左侧顶叶白质脱髓鞘;血氨基酸及酰基肉碱筛查(金域临床检验中心)显示[2-3]肉豆蔻烯酰肉碱增高,伴多种中、长链酰基肉碱增高,提示极长链酰基辅酶A脱氢酶缺乏症(verylongchainacyl-CoAdehydrogenasedeficiency,VLCADD)。尿液有机酸分析示酮尿。按VLCADD在避免劳累、避免空腹、预防感染基础上给予高碳水化合物和低脂饮食及对症处理,好转出院。
2例患儿为第二胎,平素不喜肉和油性食物。体液无特殊气味。第一胎为女孩,生后数天不明原因死亡,具体不详(家系图见图1)。2例患儿ETFDH基因发现复合杂合核苷酸变异:c.250G→A、c.498G→A,上述变异分别导致第84号氨基酸由丙氨酸变为苏氨酸(p.Ala84Thr)、第166号氨基酸由蛋氨酸变为异亮氨酸(p.Met166Ile),均为错义突变。在受检者ETFDH基因所发现的复合杂合变异分别遗传自其父母,父母均为杂合子,符合常染色体隐性遗传方式。对2例患儿及父母行遗传代谢病相关基因二代测序(北京康旭医学检验所),结果(表1、图2)显示ETFDH基因复合杂合突变,故更改临床诊断为戊二酸血症Ⅱ型。回访男性患儿,发现院外呕吐再次发作,给予补液治疗好转;更改治疗方案(增加维生素B2150mg/d口服,左旋肉碱1g/d辅助治疗)。电话随访至今,2例患儿未再有呕吐等异常发生。
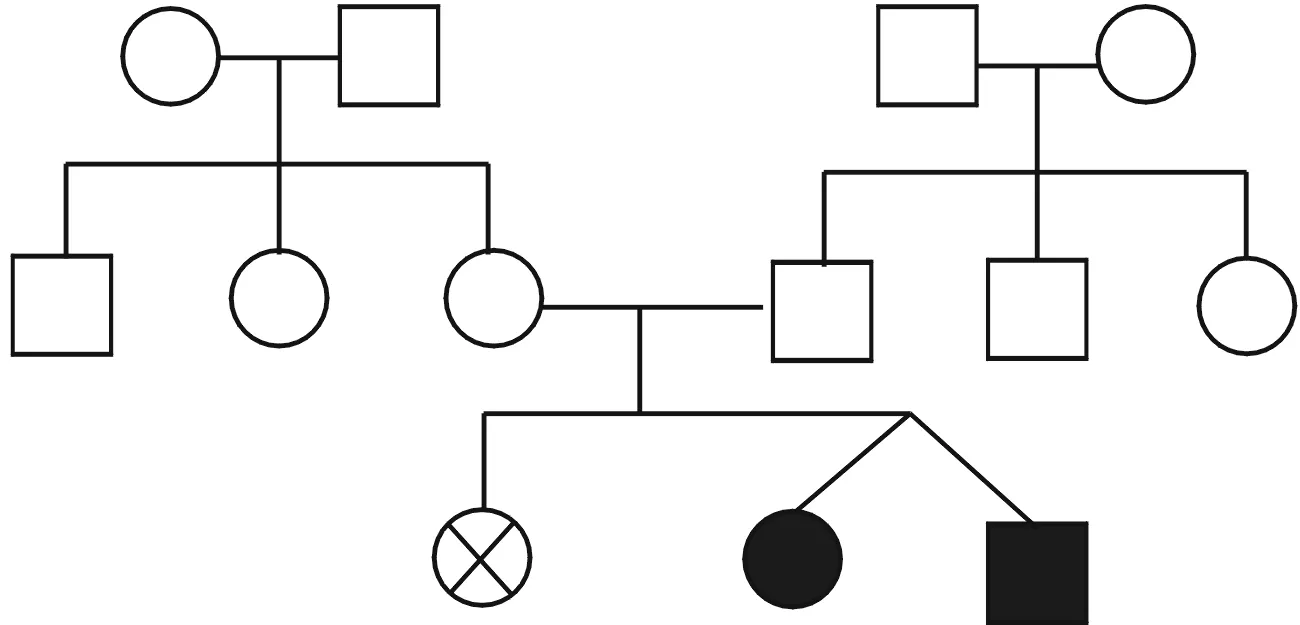
○:正常女性;□:正常男性;⊗:死亡女性;●:患病女性;■:患病男性

图1 家系图
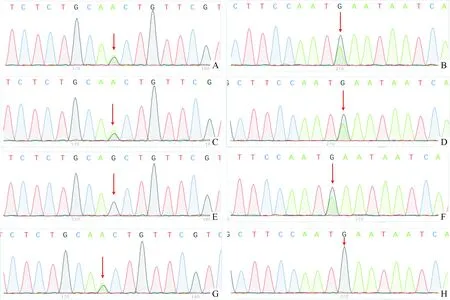
图2 双胞胎患儿及父母遗传代谢病相关基因二代测序图(图注见表1)
2 讨论
本文报道了2例误诊的戊二酸血症Ⅱ型双胞胎患儿,异卵双生,11岁。根据其临床表现、血串联质谱、尿气相质谱分析结果初步诊断为VLCADD,按VLCADD给予治疗后仍有呕吐,随后进行相关基因二代测序,结果提示戊二酸血症Ⅱ型,ETFDH基因复合杂合突变,核对测序结果,更改临床诊断为戊二酸血症Ⅱ型。两患儿均有酮尿,尿有机酸分析亦提示酮尿,与戊二酸血症Ⅱ型脂肪酸代谢障碍发病机制相悖,可能与酶的残余活性、严重呕吐、饥饿有关。根据相关基因测序报告结果,及时召回患儿继续避免感染、腹泻、饥饿等常规治疗,添加维生素B2,同时给予左旋肉碱辅助治疗,电话随访患儿未再呕吐,活动耐力明显好转,继续随访观察。
戊二酸血症Ⅱ型可分为新生儿期发病而无先天性畸形、新生儿期发病伴有先天性畸形和迟发型3种,其中新生儿期发病者病情危重,多在出生后数小时或数天出现代谢性酸中毒、低血糖、呼吸困难及高氨血症等,并常有“汗脚样”气味。迟发型患者生后数周至成人均可发病,临床表现多样且无特异性,主要为间歇性肌无力,可有心肌、肝脏受累,部分患者可在应激状态下急性发作,甚至危及生命[4]。该症的临床特点为代谢性酸中毒、高氨血症、脂质沉积性肌病,以及反复发作的非酮症性或低酮症性低血糖[5]。此病在诊断上可根据尿液中大量戊二酸、乙基丙二酸、异戊酸及多种二羧酸作为诊断依据[6],但间歇期可正常。目前基因诊断是确诊的金标准。临床疑诊患者应尽早进行血串联质谱、尿气相质谱等相关检查;基因检测不仅能够确诊,而且对其治疗和预后有指导意义。目前,对于戊二酸血症Ⅱ型尚无特效治疗方案,在饮食干预的基础上补充核黄素、辅酶Q10以及肉碱可明显改善症状和生化指标。
VLCADD是一种脂肪酸代谢障碍性疾病,为常染色体隐性遗传,根据发病年龄及临床表现分为:新生儿早发及婴儿型、儿童型和迟发型。新生儿早发及婴儿型又称心肌病型,此型最常见,且病情凶险,死亡率高,表现为低酮性低血糖、心肌病、脑病、Reye综合征。儿童型又称肝病型,表现为低血糖和异常低血酮症,肝脏肿大,几乎不累及心肌。迟发型又称肌病型,此型主要在青少年至成年期发病,临床症状轻微,主要表现为肌无力、运动不耐受以及运动或感染后引发的横纹肌溶解,较少发生低血糖[7-8]。VLCADD急性发作期可有低酮性低血糖、代谢性酸中毒、肌酸激酶等的升高,肌病型患者可有肌红蛋白尿或伴有肾功能异常,肌肉活检可发现肌肉组织中有大量脂滴蓄积于Ⅰ型肌纤维。目前诊断VLCADD最常用的方法是血串联质谱分析,其中以肉豆蔻烯酰基肉碱作为最重要的指标,若其大于1 μmol/L可诊断为VLCADD[9]。基因突变分析是确诊VLCADD的金标准[9]。VLCADD的治疗原则是避免劳累、避免空腹[10],给予高碳水化合物和低脂饮食,尤其限制长链脂肪酸的摄入,补充中链甘油三酯,对症处理及预防和治疗并发症。2例患儿为11岁,且发病年龄9岁,临床出现反复呕吐、酸中毒、酮尿等,而无心肌、肝脏、肌无力等症状,患儿在临床症状上不典型,但血尿串联质谱检测结果支持其诊断,这是导致误诊的一大原因。
戊二酸血症Ⅱ型和VLCADD均属于脂肪酸代谢障碍性疾病,可致多脏器损伤,临床表现有相似之处,如代谢性酸中毒、抽搐、喂养困难等,均可累及心脏、肝脏、肾脏及运动系统,在临床工作中不易鉴别。串联质谱、气相色谱技术是遗传代谢病初筛手段,也是新生儿筛查的主要检查[11-12]。尿或血中的代谢蓄积物分析可帮助筛选部分遗传代谢病,检查所需标本用量少、检测时间短、成本低廉,对疾病诊断具有极高的敏感度和特异度,现已在我国较广泛应用。迟发型MADD患儿尿中戊二酸、乙基戊二酸、异戊酸以及多种二羧酸等代谢物水平在疾病间歇期可正常,这给诊断带来了困难。但两患儿均有酮尿,有机酸尿液分析戊二酸水平正常,可出现阴性结果或其他非特异结果。本组2例戊二酸血症Ⅱ型的代谢筛查结果误导了对该病的及时正确诊断,提示临床表现或生化检测不完全符合或规范治疗未达到预期效果时,应及时结合基因检测,防止漏诊、误诊。
本文2例患儿基因二代测序显示ETFDH基因复合杂合核苷酸变异,均为错义突变;上述变异均可导致蛋白质功能受到影响。ETFDH是位于线粒体内膜的单体蛋白,核黄素反应性MADD患者多携带ETFDH基因突变,基因诊断为金标准。戊二酸血症Ⅱ型为常染色体隐性遗传,该症在临床中较为罕见,异卵双胞胎同时患病的概率更低,如若此患儿家属有意再孕,可结合基因突变行产前诊断。因此提高MADD认识,积极开展生化和分子生物学研究,早期明确诊断,对降低病死率和改善患者生活质量对家庭行遗传咨询有重要意义。
猜你喜欢
中国现代医生(2022年19期)2022-11-04
现代仪器与医疗(2022年1期)2022-04-19
食品安全导刊(2021年20期)2021-08-30
中老年保健(2021年4期)2021-08-22
现代仪器与医疗(2021年2期)2021-07-21
心肺血管病杂志(2020年5期)2021-01-14
科学养鱼(2020年1期)2020-03-03
分析化学(2018年12期)2018-01-22
食品界(2017年4期)2017-05-17
家庭百事通·健康一点通(2017年3期)2017-03-22
