
阿帕鲁胺合成路线图解
2019-06-26 05:58刘耀威万灵子茆勇军
山东化工 2019年11期
刘耀威,万灵子,茆勇军
(上海工程技术大学 化学化工学院,上海 201600)
阿帕鲁胺(apalutamide,1,图1),英文化学名为4-[7-[6-Cyano-5-(trifluoromethyl)-3-pyridinyl]-8-oxo- 6-thioxo-5,7-diazaspiro[3.4]oct-5-yl]-2-fluoro-N-methylbenzamide,代号为ARN-509,CAS号:956104-40-8。中文化学名为4-[7-(6-氰基-5-三氟甲基吡啶-3-基)-8-氧代-6-硫代氧基-5,7-二氮杂螺[3.4]辛-5-基]-2-氟-N-甲基苯甲酰胺。阿帕鲁胺是第2代非甾体雄激素受体(androgen receptor, AR)抑制药[1-2],自2009年开始由美国Aragon制药公司开发,该公司于2013年8月被美国强生公司收购,后者于2017年12月向美国食品药品管理局(FDA)提交新药上市申请,并于2018年2月14日获准上市,商品名为Erleada,用于治疗非转移性(前列腺癌细胞未扩散)去势抵抗(激素治疗后疾病仍进展)的前列腺癌[3]。
本文总结了1的合成路线(如图1中所示)。根据现有文献的报道,制备该化合物主要的方法包括两类:A.由N-甲基-2-氟-4-(1-氰基环丁胺基)苯甲酰胺(2)和2-氰基-3-三氟甲基-5-氨基吡啶(3)与硫光气反应环合制备得到1[4-7];B.由化合物3与硫光气反应制备得到2-氰基-3-三氟甲基-5-异硫氰基吡啶(5)[8],而后可分别与化合物2[8]、或1-((3-氟-4-(甲基氨甲酰基)苯基)氨基)环丁烷-1-羧酸甲酯(4)[10]、或1-((3-氟-4-(甲基氨甲酰基)苯基)氨基)环丁烷-1-(4-硝基苯基)羧酸酯(15)[11]反应制备得到1。其他类的方法还包括:(1)由4-(8-氧代-6-硫代氧基-5,7-二氮杂螺[3.4]辛-5-基)-2-氟-N-甲基苯甲酰胺(16)与2-氰基-3-三氟甲基-5-溴吡啶(17)反应得到1[12];(2)由4-((1-((6-氰基-5-(三氟甲基)吡啶-3-基)氨甲酰)环丁基)氨基)-2-氟-N-甲基苯甲酰胺(32)与1,1'-硫代羰基二-2(1H)-吡啶酮反应制备1[13]。(3)由4-(7-(6-氰基-5-(三氟甲基)吡啶-3-基)-8-氧代-6-硫氧基-5,7-二氮螺环[3.4]辛-5-基)-2-氟苯甲酸甲酯(34)与甲胺反应制得1[13]。下边将对上述路线作进一步阐述。
1 关键中间体的合成路线综述
1.1 N-甲基-2-氟-4-(1-氰基环丁胺基)苯甲酰胺(2)的合成
根据文献报道,制备化合物2的方法有3类:
(1)由2,4-二氟苯甲酰氯(6)与一甲胺以四氢呋喃做溶剂反应制备得到2,4-二氟-N-甲基苯甲酰胺(7)[8]收率为100%;化合物7与4-甲氧基苄胺以乙腈为溶剂,190℃下进行微波反应,制备得到2-氟-4-甲氧基苄胺-苄基酰胺甲酯(8),收率40%[8];再将化合物8与三氟乙酸在二氯甲烷为溶的条件下剂,反应制备得到4-氨基-2-氟-N-甲基苯甲酰胺(9)[8]收率为95%;最后将9与环丁酮进行Strecker反应得到2[9,11],收率为70%,总收率为26.6%。该路线中使用到微波反应条件,收率较低,并且使用4-甲氧基苄胺为保护基团,原子不经济,故该方法不具备工业化价值。
(2)将2-氟-4-硝基苯甲酸(10)悬浮在甲苯中,加入氯化亚砜酰化得到2-氟-4-硝基苯甲酰氯(11)[14]收率为100%;再将11与一甲胺反应得到2-氟-4-硝基苯甲酸(12)[14]收率为90%;再将12用铁粉还原得到4-氨基-2-氟-N-甲基苯甲酰胺(9),收率为97%[15];再按“(1)法”后续步骤制得2,4步反应总收率为52%。该方法各步反应收率均较高,虽然铁粉还原、氰化钠关环的反应具有一些不足,总体看来该方法是目前最具备工业化价值的方法。
(3)中间体12也可直接由10与一甲胺盐酸盐和N,N'-羰基二咪唑(CDI)直接化合制备得到,再按“(A)法”后续步骤制得2,该步收率为95%,3步反应总收率为64.5%。CDI缩合的方法对于工业化制备还是成本过高。
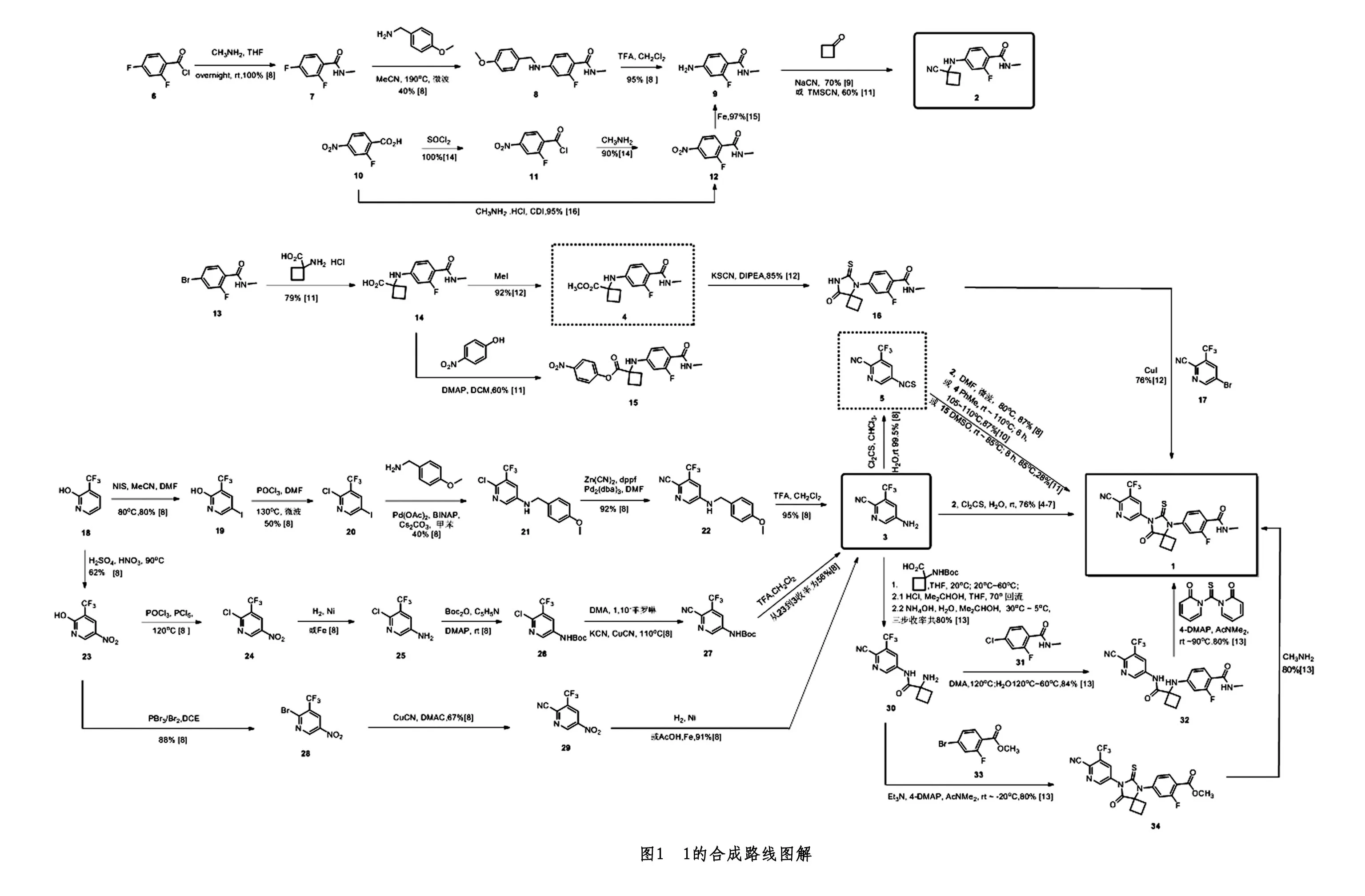
1.2 2-氰基-3-三氟甲基-5-氨基吡啶(3)的合成
根据文献报道,以2-羟基-3-三氟甲基吡啶(18)为原料制备化合物3的路线主要有3类:
(1)将化合物18溶于无水乙腈/DMF=1/1中,然后加入N-碘代丁二酰亚胺在80℃下制备得到2-羟基-3-三氟甲基-5-碘吡啶(19)[8],收率为80%;然后将19与POCl3在DMF中混合,并在微波中加热至130℃,得到2-氯-3-三氟甲基-5-碘吡啶(20)[8],收率为50%;化合物20与Pd(OAc)2、BINAP(1,1'-联萘-2,2'-双二苯膦)、Cs2CO3、4-甲氧基苄胺和三乙胺反应,得到6-氯-N-[(4-甲氧基苯基)甲基] -5-(三氟甲基)吡啶-3-胺(21)[8],收率为40%;21在氰化锌/三(二亚苄基丙酮)二钯和DMF中的双(二苯基膦基)二茂铁的溶液中反应,得到5-((4-甲氧基苄基)氨基)-3-(三氟甲基)-2-氰基吡啶(22)[8],收率为92%;22在二氯甲烷和三氟乙酸的溶液中脱去保护基,得到2-氰基-3-三氟甲基-5-氨基吡啶(3)[8],收率为95%,以上五步总收率为14%。该路线中使用到微波反应条件、使用4-甲氧基苄胺为保护基团、也使用了贵金属催化的偶联反应,生成成本过高,不具备工业化价值。
(2)将18溶于硫酸加热至90℃并加入硝酸,制备得到2-羟基-5-硝基-3-(三氟甲基)吡啶(23)[8],收率为62%;将23,POCl3和PCl5的混合物,在110~120℃下反应,制备得到2-氯-5-硝基-3-(三氟甲基)吡啶(24)[8];将24溶于四氢呋喃中,采用雷尼镍催化氢化反应,得到6-氯-5-(三氟甲基)吡啶-3-胺(25)[8];将25溶于吡啶中,并加入4-二甲基氨基吡啶(DMAP)、二碳酸二叔丁酯,得到N-6-氯-5-(三氟甲基)吡啶-3-基氨基甲酸叔丁酯(26)[8];将化合物26溶于二甲基乙酰胺(DMA)中,加入菲咯啉催化,将混合物加热至80℃并加入KCN和CuCN,在110℃下搅拌2h,得到N-6-氰基-5-(三氟甲基)吡啶-3-基氨基甲酸叔丁酯(27)[8];通过使用三氟乙酸(TFA)脱去Boc保护基团,得到(3)[8],以上6步总收率为34.7%。该路线中使用到Boc保护基团,增加了反应步骤和成本,使用氰化钾来作氰基取代试剂,生产风险很高,工业化价值不大。
(3)将化合物23用三溴氧磷进行溴化,得到2-溴-5-硝基-3-(三氟甲基)吡啶(28)[8],收率为88%;再将28溶解在二甲基乙酰胺(DMA)中,并加入CuCN和菲咯啉(0.2当量)催化,将混合物加热至160℃反应,并通过色谱法纯化得到5-硝基-3-(三氟甲基)吡啶-2-甲腈(29)[8],收率为67%;将29通过催化氢化或还原铁粉还原得到3[8],收率为91%。该路线通过硝化、溴代、氰代、还原4步反应,总收率为33.3%,路线清楚简洁、原料易得、操作方便,具备工业化价值。
2 阿帕鲁胺的合成路线综述
(1)由N-甲基-2-氟-4-(1-氰基环丁胺基)苯甲酰胺(2)和2-氰基-3-三氟甲基-5-氨基吡啶(3)与硫光气反应环合制备得到1,使用胶色谱法分离得到目标产物1[4-7],收率为76%,该路线总收率最高为22.4%。由于需要使用硫光气,其对操作人员健康危害极大并在工业化生产过程中易发生安全事故,故该路线不适合工业化大规模生产。
(2)由化合物3与硫光气反应制备得到2-氰基-3-三氟甲基-5-异硫氰基吡啶(5)[8],而后可分别与化合物2[8-9]在DMF溶液中通过在80℃微波加热反应(收率87%),路线总收率为19.4%;或1-((3-氟-4-(甲基氨甲酰基)苯基)氨基)环丁烷-1-羧酸甲酯(4)[10-11]在甲苯为溶剂中在110℃反应6 h(收率87%),总收率为19.4%;或由5与1-((3-氟-4-(甲基氨甲酰基)苯基)氨基)环丁烷-1-(4-硝基苯基)羧酸酯(15)在DMSO中控温85℃反应制备得到1[11](收率为28%),化合物15可由14与4-硝基苯酚在4-二甲氨基吡啶(DMAP)和N,N'-二环己基碳酰亚胺(DCC)中制备得到[11],收率为60%。上述三条路线最高总收率为19.4%。
(3)由4-溴-2-氟-N-甲基苯甲酰胺(13)与1-氨基-1-环丁烷羧酸盐酸盐化合制得1-((3-氟-4-(甲基氨基甲酰基)苯基)氨基)环丁烷-1-羧酸(14)[11],收率为79%;14和碘甲烷制备得到1-((3-氟-4-(甲基氨基甲酰基)苯基)氨基)环丁烷-1-羧酸甲酯(4)[12],收率为92%;4与N,N-二异丙基乙胺和氰化钾反应制备得到4-(8-氧代-6-硫代氧基-5,7-二氮杂螺[3.4]辛-5-基)-2-氟-N-甲基苯甲酰胺(16)[12],收率为85%;16与2-氰基-3-三氟甲基-5-溴吡啶(17)反应得到1[12],收率为76%,该路线总收率为47%。该路线各步反应收率均较高,反应条件温和、操作方便,是目前具备放大潜力的路线。
(4)由3与1 -((叔丁氧基羰基)氨基)环丁烷-1-羧酸,经缩合、脱保护、游离3步反应,制备得到1-氨基-N-(6-氰基-5-(三氟甲基)吡啶-3-基)环丁烷-1-甲酰胺(30)[13],总收率为80%,30再与4-氯-2-氟-N-甲基苯甲酰胺(31)反应制备得到4-((1-((6-氰基-5-(三氟甲基)吡啶-3-基)氨甲酰)环丁基)氨基)-2-氟-N-甲基苯甲酰胺(32)[13],收率为84%;再将32与1,1'-硫代羰基二-2(1H)-吡啶酮反应制备1[13]收率为80%,总收率为17.9%。
(5)由30与4-溴-2-氟苯甲酸甲酯(33)进行反应得到4-(7-(6-氰基-5-(三氟甲基)吡啶-3-基)-8-氧代-6-硫氧基-5,7-二氮螺环[3.4]辛-5-基)-2-氟苯甲酸甲酯(34)[13],收率为80%,再将34与甲胺反应制得1[13],该路线总收率为22.2%。
猜你喜欢
浙江化工(2022年8期)2022-09-05
河南化工(2021年4期)2021-05-12
昆明医科大学学报(2021年2期)2021-03-29
农药科学与管理(2019年10期)2019-04-20
文化产业(2016年6期)2016-10-19
纺织学报(2016年3期)2016-05-25
中国塑料(2016年11期)2016-04-16
中国粮油学报(2016年1期)2016-02-06
浙江化工(2015年4期)2015-11-28
郑州大学学报(理学版)(2014年4期)2014-03-01
