
一例1q23.3-q25.2缺失综合征型耳聋报道及文献复习
2019-10-24 05:18张飞牛文侠丁韶洸谢存存贾晓东秦利涛刘宏建
中华耳科学杂志 2019年5期
张飞牛文侠丁韶洸谢存存贾晓东秦利涛刘宏建*
1郑州大学附属儿童医院、河南省儿童医院、郑州儿童医院耳鼻咽喉科(郑州450053)
2河南大学人民医院(郑州450003)
3河南省人民医院耳鼻咽喉科、郑州大学人民医院、河南大学人民医院(郑州450003)
4郑州大学人民医院、河南省人民医院、河南省医学遗传研究所(郑州450003)
遗传性耳聋是人类常见的遗传疾病之一,患儿出生时约1∕1000人中伴有听力障碍的先天性耳聋患者[1]。遗传性耳聋具有遗传质异性,30%为综合征型耳聋,常见为Usher综合征、Pendred综合征等,70%为非综合征型耳聋[2],多见于GJB2、GJB3、SLC26A4基因引起。而1号染色体长臂缺失(1q)引起的综合征型耳聋却较为罕见,本研究采用微阵列比较基因组杂交(array comparative genomic hybridization,aCGH)技术报道一例1号染色体q23.3-q25.2臂间缺失的综合征型耳聋患者,并复习相关文献,提高对该疾病的认识。
1 病例资料
患者,女,7个月,因双耳听力差就诊。患儿产前羊水过少,足月顺产,出生时体重2.3 Kg(-3SD为2.26 kg),身高44 cm(-2SD为44.7 cm),头围25 cm(-3SD为30.4 cm),出生后3次听力筛查未通过。现患儿体重5 Kg(-3SD为5.90 kg),身高56 cm(-3SD为61.3 cm),头围36 cm(-3SD为39.5 cm[3]),不能翻滚,精神运动迟缓。查体:头发及眉毛稀疏,五指短小、第五手指弯曲,鼻梁较宽,右侧杯状耳、副耳,左侧垂耳、小耳(图1)。
听力学检查:气导ABR检查双耳V波反应阈值均为55 dBnHL,骨导ABR左耳V波反应阈值为35 dBnHL,右耳V波反应阈值为30 dBnHL,ASSR双耳平均阈值60 dB,耳声发射双耳未通过,声导抗双耳“A”型曲线(图2)。
影像学检查:颞骨CT检查未见明显异常(图3);超声检查显示心脏彩超提示卵圆孔未闭;肾脏彩超提示双肾脏体积偏小。
生化检测:出生3月时肌张力低下,检测发现维生素B12缺乏,检查值为153.854 pg∕mL(参考范围200~900 pg∕mL)。补充维生素B12恢复正常后,患儿肌张力恢复正常。
遗传咨询:患儿父母非近亲,无类似家族史(家系图见图4A)。采用aCGH技术检测,抽取质控合格的患儿及父母外周血基因组DNA 500 ng,使用Sure-Print G3 Human CGH Microarray Kit,8x60K G4450A(Agilent公司,美国)芯片进行检测,SureScan Microarray Scanner扫描系统对探针信号进行扫描,Agilent CytoGenomics 2.9软件对扫描结果分析,设置对连续三个探针log2值≥0.25或log2≤-0.25且≥200 kb染色体片段重复、缺失部分进行分析,结果提示arr[hg19]1q23.3-q25.2(161316029-178987393),即1q23.3-q25.2区存在17.67 Mb杂合缺失;父母结果提示无相同片段染色体异常(图4B)。
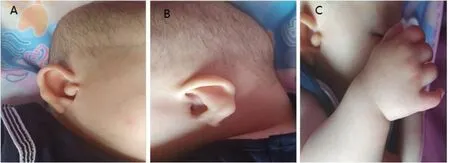
图1 A右耳:杯状耳、副耳;B左耳:垂耳、小耳;C五指短小,第五手指弯曲.Fig.1 Clinical manifestation:The right ear was cup-shaped and accessory ear(A);B,The left ear was characterized by lopsided ear and small ear(B);The hand was characterized by short five fingers and the fifth finger curved(C)

图2 A患儿听力学检测(2019-1-14,我院):气导ABR检测显示双耳V波均为55dBnHL;B骨导ABR显示左耳V波为35dBnHL,右耳V波为30dBnHL;C耳声发射显示双耳未通过测试;D ASSR双耳平均阈值60dBFig.2 Audiometry detection(2019-1-14,Our Hospital):the air guided ABR detection showed that both binaural V waves were 55dBnHL(A);the bone guide ABR showed that V wave of left ear was 35dBnHL and V wave of right ear was 30dBnHL(B);the otoacoustic emission shows that both ears failed the test(C);the detection showed that binaural mean threshold of 60dB(D)

图3 外耳、中耳、内耳结构正常Fig.3 Temporal bone CT showed the outer ear,middle ear and inner ear were normal

图4 患儿家系图(A)和比较基因组杂交结果(B)。结果显示患儿(P:先证者)1q23.3-q25.2区存在17.67Mb缺失;患儿父母(F:父亲;M:母亲)不携带该缺失。注:图B纵坐标表示位于基因组中的位置,横坐标表示探针信号的log2Ratio值。每个点代表1个探针检测位点值,在没有基因组拷贝数变异的区域,信号点均匀密集的分布在基线左右,在缺失区域里信号点明显向左偏移(P,红色箭头)。Fig.4 Family diagram of the patient(A)and array-based comparative genomic hybirdyzation(array-CGH)(B).The results showed a 17.67Mb deletion was located in the region q23.3-q25.2 of chromosome 1 of the proband(P:proband);Parent(F:father;M:mother)did not carry the deletion.Note:vertical axis and horizontal axis respersent the position in the genome and the log2 ratio value of the probe signal,respectively in Fig.B.Each point represents one probe detection site value.Signal points are uniformly and densely distributed around the baseline in the region without the deletion,while signal points are significantly shifted to the left(P,red arrow)in the deletion region.
2 讨论
1号染色体长臂缺失是一种罕见的疾病,可由遗传性或自发突变引起。1977年Schwanitz等[4]首次报道1号染色体长臂缺失,分为3种类型:1 q近端缺失(1q21-22→ q 25)、1q中间缺失(1q24-25→q 32)以及1 q远端缺失(1 q42→末端)。近年来,Chatron等[5]总结该病常见临床表现为产前和产后生长迟缓、智力低下、精神运动迟缓、内分泌缺乏(甲状腺激素、生长激素、抗凝血酶III)、头颈部发育异常(小头、头发及眉毛稀疏、宽鼻梁、唇腭裂、上唇薄、短人中、小颌、短颈)、四肢畸形(五指短小、第五手指弯曲)、肾脏(马蹄肾)和心脏异常(卵圆孔未闭)、外生殖器异常(阴茎短小、隐睾)、疝气(腹股沟疝、脐疝)、耳廓异常和听力损失。
由于aCGH技术对全基因组进行扫描,可以检测染色体内微小缺失、重复等不平衡性的重排,国际细胞基因组芯片标准协作组(International Standards for Cytogenomic Arrays Consortium,ISCA Consortium)推荐将aCGH作为对原因不明的发育迟缓、智力低下、多种体征畸形以及自闭症患者的临床首选检测方法[6]。本文通过aCGH技术检测患儿为1q23.3-q25.2区17.67 Mb杂合缺失,属于1q近端缺失,由于患者父母无相同片段染色体异常,提示该患儿1q长臂缺失是自发突变引起。对于1q区域缺失引起的听力下降,早在1996年Fagerheim等[7]首次报道了DFNA7基因(OMIM:601412),定位于1q21-q23,为常染色体显性遗传,临床特征以高频开始,渐进性感音神经性聋。随后,2009年Catelani等[8]研究表明1q23.3-q25.2片段可能含有与听力相关的剂量敏感基因,且DFNM1基因(OMIM:605429)位于1q24已证实与听力障碍有关[9]。文献检索发现有10例报道1q22-q25.2区缺失的患者存在听力损失,这些区域的缺失与1q23.3-q25.2部分重叠,在一定程度上可以解释该患儿听力损失的原因(表1)。
在OMIM数据库中检索发现,CENPL基因位于1q25.1(1:173799549),编码着丝粒蛋白L,该基因的缺失会破坏着丝粒,导致细胞有丝分裂异常,造成生长异常和小头畸形[13]。研究发现,1q近端缺失与心脏、肾脏畸形也有相关[5]。本文经aCGH技术检测出该患儿包含以上基因的缺失,这些可能引起该患儿的身材矮小、小头畸形、卵圆孔未闭和肾脏体积小的临床特征。而维生素B12缺乏本身会引起生长缓慢、表情呆滞、智力下降等神经系统症状,该患儿补充维生素B12恢复正常后,患儿肌张力恢复正常,说明该患儿生长发育迟缓更可能与1号染色体q23.3-q25.2区17.67 Mb杂合缺失有关。
CENPL虽然有着一定的功能,但目前仍在研究,OMIM数据库中仍未定义临床表型编码。而OMIM数据库显示该患儿1q23.3-q25.2区(161316029-178987393)缺失的17.67 Mb包含了84个已知致病基因编码和对应的临床表型编码(表2),分别位于1q23.3、1q241、q24.1、1q24.2、1q24.3、1q25、1q25.1断裂热点。其中,位于1q23.3的PBX1、CAKUHED基因(OMIM:176310)与先天性肾脏和泌尿道异常,伴有或不伴有听力丧失、耳廓异常或发育迟缓相关[15];位于1q24的DFNM1基因(OMIM:605429)与非综合征型耳聋相关[9];位于1q24.1的TMCO1、CFSMR基因(OMIM:614123)与颅面畸形、骨骼异常和智力低下相关[16]。推测,这些片段的缺失更能说明该患儿突变致病的可能性较大。由于该区域内包含基因数目较多,这些基因的变异共同导致临床表型,其中某些基因可能会决定临床表型的方向,但目前这些关键基因尚不明确。除此之外,临床表型的差异还可能与染色体断裂位点有关,染色体的断裂缺失会引起染色质的空间构象改变,而后者与基因的表达密切相关。同时,片段缺失大小包含丢失编码基因的种类和数量不同,也会影响临床表型。因此,文献检索中有2例共同区域缺失的患者,分别为Chatron等[5]报道的1q23.3-q25.2区14.1 Mb杂合缺失,Catelani等[8]报道的1q23.3-q25.2区14.06 Mb杂合缺失,这两例患者与本文报道的临床表型相似(表1),但仍存在差异。

表1 1q22-q25.2区缺失患者听力损失的临床特征Table 1 Clinical characteristics of hearing loss in patients with the deletion of 1q22-q25.2

表2 1q23.3-25.2区缺失的基因与临床表型相关性Table 2 1q23.3-25.2 deletion of disease-related genes
总之,本文通过aCGH技术检测出患儿存在1q23.3-q25.2区的缺失,该区域的已知基因功能可以部分解释患儿综合征型耳聋的临床特征,然而该区域基因的研究尚不充足,有些基因仍未鉴定出编码功能,还需要做进一步研究。由于该病发生机制尚不明确,临床遗传咨询建议再次妊娠时做好产前诊断,降低再发的风险。同时,该病例提示我们应该重视患有发育迟缓、体形异常的患者,及早行遗传学(aCGH)和临床相关检查。
猜你喜欢
中国现代医生(2022年21期)2022-08-22
家庭科学·新健康(2022年1期)2022-02-02
家庭科学·新健康(2021年11期)2021-11-23
中医眼耳鼻喉杂志(2021年1期)2021-07-22
天津医科大学学报(2021年1期)2021-01-26
中国生殖健康(2020年4期)2021-01-18
紫禁城(2020年5期)2021-01-07
医药前沿(2020年20期)2020-11-10
三农资讯半月报(2020年2期)2020-03-09
家庭百事通·健康一点通(2019年8期)2019-08-29
