
多肽/微型蛋白类导向分子的研究进展
2019-12-23 04:03汪小又刘鑫龙李翀
药学进展 2019年10期
汪小又,刘鑫龙,李翀
(西南大学药学院,重庆 400715)
导向分子,又被称为功能性配体,主要通过识别其分布于某些细胞表面的对应受体来介导药物更有效地蓄积于靶部位。其介导药物的方式主要包括:1)配体与小分子药物形成偶联物(liganddrug conjugates);2)配体修饰于药物载体表面构建靶向递药系统(targeted drug delivery systems)。蛋白-蛋白相互作用在生命体活动中占据了主导地位,其本质则是多肽片段之间的相互识别,因此,多肽往往具有不亚于蛋白的靶受体高亲和力;同时,多肽相对分子质量较小,组织渗透性强,合成与修饰的技术成熟且生产成本相对较低,这些优势使得多肽成为得到最为广泛研究和开发的一类导向分子。目前,一些多肽-药物偶联物(peptide-drug conjugates)和多肽介导的靶向递药系统已经进入临床实验的不同阶段[1-3](见表1)。
多肽类导向分子的研究主要包括:1)针对感兴趣的靶受体、靶细胞乃至靶组织,如何获得对应的多肽导向分子;2)在已有多肽的基础上如何进一步优化以更好地胜任导向分子的“角色”。与直接发挥药效的多肽药物略有差异的是,多肽导向分子需长时间暴露于生理环境下以持续发挥靶向功能,其稳定性、潜在免疫原性等性质尤为值得关注。此外,导向分子的提出最初主要源于肿瘤靶向治疗的需求,目前已逐步拓展到许多重要疾病治疗领域。本文对近年来多肽类导向分子在来源、优化以及应用方面的新进展进行综述。
1 多肽类导向分子的来源新进展
天然发现、分子文库筛选和化学设计构成了多肽类导向分子的3类主要来源。近年来,经天然发现的可用作导向分子的多肽种类正在不断增加;对现有多肽文库进行优化(上游)和提高对筛选多肽的分析、鉴别效率(下游)提高了靶蛋白优质多肽配体的获取效率;同时,计算科学的突飞猛进使得多肽、蛋白的化学设计研究正在发生革命性的变化。

表 1 含有多肽导向分子的部分在研药物Table 1 Some peptide targeting molecule-containing drugs under development
1.1 天然发现的多肽类导向分子
自然界向药物设计工作者提供了大量具有丰富三维结构以及较强生物活性的多肽分子,这同样也构成了多肽类导向分子来源的“宝库”[4]。特别是来源于天然生物毒素、富含二硫键的多肽(具有2对及以上的二硫键)。这类多肽因多对二硫键的连接和固定而具有了高级结构,尽管其氨基酸数量相对较少(一般不超过50个),也常被称为微型蛋白(miniature protein)。这类多肽/微型蛋白与靶受体结合能力强,生物稳定性较之于线性多肽大幅提升,近年来作为潜在的导向分子受到广泛关注。
蜂毒明肽(apamin)是目前已知氨基酸数量最少的、能穿透血脑屏障(blood-brain barrier, BBB)的富二硫键多肽毒素。其具有18个氨基酸,经2对二硫键连接后具有经典的ɑ螺旋结构。小电导钙激活型钾通道蛋白1-3(small conductance calciumactivated potassium channel,SK1-3)是其受体。鉴于脊髓损伤(spinal cord injury,SCI)在临床上几乎无药可治且往往导致肢体严重的功能障碍,造成患者长期的身心严重伤害与巨大的经济负担,笔者课题组以蜂毒明肽为导向分子,构建了脊髓靶向的聚合物胶束递药系统,研究发现,蜂毒明肽同样能够高效通过血-脊髓屏障,介导药物蓄积于损伤部位,实现了较好的治疗目标[5]。Oller-Salvia等[6]对蜂毒明肽中涉及脑靶向性、细胞毒性、蛋白酶抗性的序列进行了分析和研究,在此基础上进一步构建了性能更好的衍生肽MiniAp-4作为介导脑部靶向递药的导向分子。
与蜂毒明肽相比,蝎氯毒素(chlorotoxin)的体积要大得多,其共有36个氨基酸和4对二硫键。蝎氯毒素靶点为胶质瘤细胞表面高度表达的氯离子通道-3(choloride channel-3,CLC-3),基质金属蛋白酶2(matrix metalloproteinase-2,MMP2)和annexinA2等,对神经胶质瘤具有良好选择性,因此蝎氯毒素已被设计为携带131I用于治疗恶性胶质瘤和黑色素瘤的罕见病药物,并获得美国FDA批准[7]。Díaz-Perlas等[8]开发了基于蝎氯毒素的肿瘤靶向递药系统和辅助诊断及手术的影像学探针。来自海洋腹足纲软体动物芋螺的芋螺毒素(conotoxin)是二硫键密度最高的一类多肽。Mei等[9-10]以ɑ7烟碱乙酰胆碱受体(alpha7 nicotinic acetylcholine receptor,ɑ7-nAChR)的配体 ——ɑ-conotoxin ImI为导向分子,分别构建了能高效靶向非小细胞肺癌A549和乳腺癌MCF-7的递药系统。
1.2 多肽文库技术
部分情况下,目标受体缺乏适宜配体,或已有配体不适合直接用作导向分子,此时可利用文库技术寻找适宜的导向分子。通过生物或化学的手段构建大容量的随机多肽库,不仅可筛选出针对特定靶蛋白的多肽配体,还能进一步拓展到针对特定靶细胞乃至靶组织的多肽导向分子。发展至今,多肽文库技术的种类已趋于稳定,包括基于生物的噬菌体展示肽库、细菌表面展示、核糖体展示、mRNA展示等和基于化学的一珠一化合物(one-bead-onecompound)技术等。近年来,研究者们对筛选过程的上、下游给予了更多关注,在上游优化多肽文库,从源头上提高所筛多肽的性能,在下游提高鉴别候选多肽的效率和速度以大幅减少费用和时间成本。
1.2.1 多肽文库的优化 经常规文库筛选出的大多为线性多肽和单二硫键环肽,生物稳定性不足,一些单二硫键环肽也易在病理微环境下被还原成线性小肽,后续应用受到很大限制。基于富二硫键多肽稳定性强且具有丰富的高级结构,Crook等[11]构建了一个半胱氨酸密集型多肽文库。该文库建立在哺乳动物细胞表面(称为mammalian cell display),充分发挥了哺乳动物细胞能表达/分泌种类丰富(超过1 400种)的富二硫键蛋白模板(scaffold)的功能,与之对应的是,常用来展示线性多肽的大肠埃希菌(E. coli)或酿酒酵母(S. cerevisiae)因仅能表达/分泌约不到50种的富二硫键蛋白而无法用于构建这种富二硫键多肽文库。
噬菌体展示双环肽库也是近年来涌现出的多肽文库优化新思路。双环肽具有2个氨基酸序列环,中间由一个连接体进行调控。与单环肽相比,双环肽的结构受到更多的约束,因此其对靶亲和力往往更强,对蛋白酶的耐受力也更强。双环肽文库的构建主要是通过在含有3个半胱氨酸的直链肽库上引入能与巯基反应的连接体,连接体同时与3个巯基共价连接后就形成了双环结构(见图1)。目前,双环肽文库已被用于人表面生长因子受体2(Her2)、尿激酶型纤溶酶原激活因子、凝血因子ⅩⅡa等多个疾病相关蛋白的多肽配体筛选中,所得多肽配体对靶亲和力有的已达到皮摩尔水平,展示出良好的应用前景[12-14]。
1.2.2 多肽文库筛选的新策略 众所周知,天然多肽大多由天然L型氨基酸(除甘氨酸无旋光异构体)组成,也被称为L型多肽,易被体内蛋白酶识别并降解;而由L型氨基酸的对映异构体——D型氨基酸所组成的D型多肽难以被蛋白水解酶识别,具有良好的生物稳定性。然而,通过多肽文库直接大量筛选全D型多肽并非易事:通过化学合成大容量的全D型多肽库,其成本远远高于L型多肽的获得,而基于生物的噬菌体展示、细菌展示等文库技术,因无法表达非天然的D型多肽序列而不能用于直接筛选D型多肽配体。
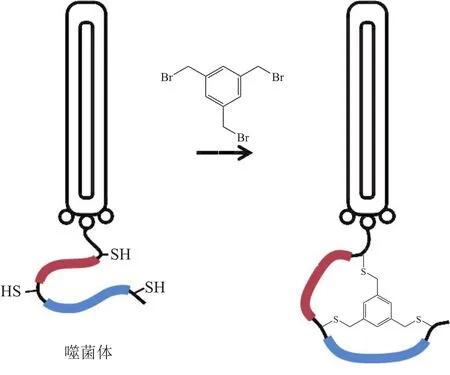
图 1 噬菌体双环肽文库的构建Figure 1 Construction of a phage bicyclic peptide library
美国麻省理工学院Schumacher等[15]提出了镜像噬菌体展示肽库的技术,其步骤为:首先通过化学全合成得到天然靶蛋白(L型)的镜像分子——D型靶蛋白,然后利用噬菌体肽库筛选得到能特异性结合D型靶蛋白的配体(L型多肽),最后合成所得配体的镜像分子——D型多肽。由于L型多肽配体能特异性结合D型靶蛋白,基于镜像对称关系,D型多肽配体也能特异性结合L型靶蛋白,故该D型多肽为天然靶蛋白的镜像配体。这一工作与其说是一种新技术,不如说是一种新策略,是在未改变常规多肽文库的前提下,仅通过对靶蛋白和配体的依次构型转换就实现了D型多肽的高效筛选,所得的D型多肽配体特异性好、稳定性强。这一策略先后在抑制原癌蛋白(p53-MDM2)、弱化HIV侵袭力(gp41外膜蛋白)和阻止β-淀粉样多肽自聚等方面的D型功能多肽筛选上得到成功的应用[16-18]。成纤维细胞生长因子诱导分子-14(Fn14)是近年来发现的胰腺癌新靶标,更有意义的是,Fn14也在肿瘤相关成纤维细胞上高表达。Fn14的胞外区长50个氨基酸,含有3对二硫键,笔者课题组合成了全D型的Fn14胞外区并基于镜像噬菌体肽库的策略筛选得到针对Fn14的多肽配体,这也是首个报道的经镜像噬菌体展示得到的多肽导向分子[19-20]。针对难渗透型的胰腺癌BxPC3模型,该D型多肽介导脂质体药物能够高效靶向病灶并能有效渗透到瘤深处,表现出良好的疗效。
1.2.3 候选多肽高效分析鉴定 新一代测序技术(next-generation sequencing,NGS)在基因组装配、基因表达定量和元基因组分析等领域得到广泛应用。因其功能强大,近几年来也逐渐被应用于噬菌体肽库测序。传统Sanger测序是劳动密集型,仅限于鉴别少于102个,极少数情况下约103个配体。相反,NGS平台能够一次表征高达106~ 108个序列。't Hoen等[21]以噬菌体七肽肽库筛选针对KS483成骨细胞的多肽导向分子,并以此为例系统比较了传统测序方式和NGS在噬菌体展示上的区别。研究结果发现,基于新一代技术的深度测序极大地简化了对共有基序(consensus motifs)的鉴定,仅需一轮筛选即可,从而避免了需要反复轮次的筛选和扩增,也能通过检测低丰度的克隆来改善配体选择的效率并极大地阻止了假阳性结果的产生。
1.3 多肽的化学设计
多肽的化学设计通常并不能和天然多肽蛋白割裂开来,这是因为大多经化学设计获得的多肽仍来源于天然配体或蛋白-蛋白相互作用的结构域。往往在已有序列和结构的基础上,计算机化学能够辅助确定关键氨基酸和作用区域(氨基酸长度)等信息,并能在一定程度上指导对所得多肽的进一步优化。Zhan等[22]通过计算机辅助设计从蛇毒蛋白candoxin中第2个loop区域中截取了长为16个氨基酸的多肽,该多肽保留了candoxin能与BBB上α7-nAChR特异性结合的功能,能够作为介导脑靶向递药的导向分子。
表皮生长因子受体(EGFR)广泛表达于肺癌、乳腺癌、膀胱癌等多种癌细胞表面,是重要的抗肿瘤靶标之一。然而,以EGFR的抗体为导向分子介导载药系统抗肿瘤的表现并不理想,为此,Song等[23]基于Mekler-Idlis氨基酸配对理论,以EGFR结构域1(domain I)为靶点设计了132条小肽配体并从中优选出序列为Leu-Ala-Arg-Leu-Leu-Thr的六肽。以该多肽为导向分子能实现对EGFR高表达的非小细胞肺癌H1299体外和体内的高效靶向。华盛顿大学的Baker等[24-26]近年来专注于微型蛋白的化学设计,他们在已有的富二硫键多肽/微型蛋白基础上引入非经典结构,丰富了具有良好热稳定性和耐酶降解的模板库,在此基础上,他们构建了数量高达2万种以上、经从头设计(de novodesign)的微型蛋白虚拟库,先后在流感病毒A H1血凝素、白细胞介素(IL)2受体等靶蛋白上进行了配体的筛选,通过化学设计结合实验验证,得到的微型蛋白配体不仅能与相应受体高度特异性结合,同时还表现出高度稳定性以及较低的免疫原性。
计算及信息科学的快速发展也为多肽导向分子的化学设计带来新的理念。与传统依靠经验设计的方法不同,Manavalan等[27]开发了一种基于机器学习多层次筛选细胞穿膜肽(cell-penetrating peptide,CPP)的预测方法。细胞穿膜肽能够携带生物大分子乃至递药系统进入细胞内,也是一类重要的多肽导向分子。通过计算包括氨基酸组成、二肽组成、氨基酸指数、成分转变-分布系数和理化性质等信息,该方法首先可预测未知肽是否是CPP,其次可以预测细胞对于该肽的摄取效率。其所创建计算方法的筛选精确率达72.5%,明显优于其他现行的CPP预测方法。为了方便科研工作者使用,作者还设计了可公开访问的web服务器:www.thegleelab.org/MLCPP。
2 多肽类导向分子优化方法与策略的新进展
在获取多肽导向分子的初始阶段中,多肽与靶标的亲和力和选择性是最受关注的目标。尽管如前所述,多肽文库或化学设计技术的进步已从源头上提升了初筛多肽的质量,但不可否认的是,线性小肽仍是初筛阶段的主要产物。这就需要研究者在兼顾亲和力的同时,对候选多肽进行优化以满足复杂生理环境下的应用需求。本部分主要对多肽导向分子的稳定性、免疫原性和功能集成3个方面的优化研究进行探讨。
2.1 稳定性优化
稳定性是多肽导向分子发挥功能的前提。复杂多变的病理生理环境如多种水解酶的水解以及pH变化等,会使多肽导向分子结构断裂、构象改变并被迅速清除,难以持续发挥靶向功能。为了维持导向分子结构和功能的稳定,研究者们近年来发展了嫁接、硒化、逆序、订书肽等多种技术与方法。
2.1.1 硫-硒环化策略 多肽分子环化的研究从上世纪就已经开始,是一种经典的稳定性优化策略。但长期以来的单环化策略大多仅是将多肽的刚性增强,有时还会导致亲和力的损失,部分单环的稳定性也不十分理想。近年来,以富二硫键多肽为模板(scaffold)来实现线性多肽的环化成为研究热点。这一过程大致可分为:1)确认候选多肽的关键氨基酸位点和受体结合态结构,线性多肽在游离状态下一般结构无序,但当其与靶受体结合时会形成相应的高级结构;2)选择与候选多肽受体结合态结构相似的富二硫键多肽,作为模板;3)将候选多肽的关键氨基酸“嫁接”到模板肽上。新生成的多肽保留了原有线性多肽的功能并由于多对二硫键的固定,其稳定性大幅提高。更值得一提的是,新生成的多肽由于提前形成了活性构象,避免了从无序到有序变化的熵消耗,因而其对靶受体的亲和力也得到了补偿式的提高(与原线性多肽相比),很好地兼顾了稳定性和功能性。笔者课题组曾以部分肿瘤细胞膜表面和胞内均高表达的原癌蛋白鼠双微粒体MDM2、MDMX为靶点,将p53与其结合的关键位点嫁接到蝎毒素BmBKTx1的α螺旋中,得到了能够有效抑制p53与MDM2/MDMX 结合的微型肽[28]。另外,同样针对该靶点,经噬菌体展示筛选出了线性多肽配体PMI(序列为:TSFAEYWNLLSP),通过端基截取以及丙氨酸扫描等方法发现PMI与MDM2结合的活性位点分别为第3位苯丙氨酸(Phe3)、第6位酪氨酸(Tyr6)、第7位色氨酸(Trp7)和第10位亮氨酸(Leu10),而PMI与MDM2结合时其构象为α螺旋结构。课题组以蜂毒明肽为模板进行嫁接,由于其α螺旋结构始于第9位丙氨酸,嫁接方法为将其Ala9/Ala12/Arg13/Gln16分别用Phe/Tyr/Trp/Leu取代。嫁接肽具有良好的靶结合能力,且其血清半衰期达到原直链短肽的近100倍[29]。
肿瘤等部分疾病存在的还原性病灶微环境使得经二硫键加固的环肽仍有进一步提高稳定性的需求。一些研究者采用了更为稳定的硒硫键或二硒键代替二硫键以增强多肽对还原性环境的耐受能力。这个过程大多采用的是硒代半胱氨酸,也就是自然界第21种天然氨基酸来取代半胱氨酸。Arai等[30]将胰岛素分子中半胱氨酸二硫键(S-S)替换为硒硫键(Se-S),以获得具有较长半衰期的胰岛素衍生物。硒化胰岛素(Se-Ins)维持了与牛胰岛素类似的活性,且在胰岛素降解酶(insulin degrading enzyme, IDE)的作用下,降解速度明显减慢(Se-Ins降解半衰期约8 h,牛胰岛素降解半衰期仅为约1 h)。笔者课题组将经二硫键连接的肿瘤靶向环肽Lyp-1进行硒化修饰,得到硒化类似物Syp-1。表面等离子共振结果证明 Syp-1保持了与靶受体p32的特异性结合能力,并较之Lyp-1表现出显著增强的血清稳定性以及更优的体内外效果,Syp-1修饰的载阿霉素脂质体对 MDA-MB-435 的体外 IC50为 588 nmol·L-1,与 Lyp-1(1.48 µmol·L-1)相比显著降低,在体内抑瘤试验中也有相同趋势[31]。
2.1.2 逆序策略 逆序策略,也称为逆反化(retroinverso isomerization),是指将某些常规多肽的L型氨基酸全部替换成D型氨基酸,并将氨基酸序列颠倒,这样得到的D型多肽与原来的多肽具有相似的侧链拓扑学结构,因而可以替代原来的L型多肽。这种策略可以根据已有的L型多肽快速获得具有高度稳定性的全D型多肽。BBB是药物递送入脑的重要生理屏障,BBB上的乙酰胆碱受体(nAChR)等受体介导的穿细胞通路是跨越该屏障的重要途径,然而该穿细胞转运通路涉及溶酶体,极易对相关导向分子造成酶解,影响其递送效果。因此,要实现有效的跨BBB转运,需要导向分子在溶酶体中稳定,这对导向分子的稳定性提出了很高要求。Wei等[32]在其前期通过化学设计获得nAChR特异性结合的L型多肽配体CDX的基础上,经逆序设计获得了对应的D肽分子DCDX。较LCDX而言,DCDX不仅提升了L型肽原有的活性,还将其血清稳定性提高了近50倍,同时还大大提高了其在胞内溶酶体中的稳定性,使其在整个递送过程中维持活性,有效跨越BBB。将DCDX修饰于脂质体表面后,与原有L型肽相比,其在脑胶质瘤模型上表现出更强的脑靶向能力和显著更优的治疗效果。值得注意的是,逆序策略并非能任意使用,例如活性结构为α螺旋结构的多肽就不适用。逆序策略可以和筛选D型多肽的镜像噬菌体展示技术在一定程度上互补[33-34]。
2.1.3 订书肽策略 与将多肽分子进行环化而提高结构刚性不同,订书肽技术(peptide stapling)旨在保护多肽结构中α螺旋三级结构。α螺旋是蛋白类分子常见的结构,其中,在蛋白-蛋白相互作用区域的α螺旋结构对蛋白类分子的活性和功能常有重要意义。然而许多较短的多肽和微型蛋白类分子中,由于缺乏其他部位空间结构的支撑和限制,其α螺旋结构通常难以稳定维持其原本的构象和活性。因此,研究者们选用处于α螺旋中同一平面的2个氨基酸进行订合,从而对该α螺旋结构进行加固。目前的多肽订合反应可分为2种:1)单组分订合法:同一多肽分子中不同氨基酸侧链直接键合;2)双组分订合法,通过一个具有双功能的连接物,对不同氨基酸侧链进行桥联(见图2)。

图 2 单组分和双组分订合法Figure 2 One- and two-component stapling
Dietrich等[35]使用订书肽策略设计了针对β-连环蛋白和T细胞因子/淋巴细胞增强-结合因子转录因子之间相互作用的抑制剂,从而抑制肿瘤相关的Wnt信号通路。同时,为了使目标多肽能进入细胞并达到作用部位,作者借鉴了部分穿膜肽疏水、带正电的特点进行优化,最终使用订书肽策略设计的NLS-StAx-h蛋白-蛋白相互作用抑制剂具有较好的选择性和较高的稳定性,还具有较高的细胞摄取量,能够有效抑制肿瘤Wnt信号通路相关的生长和迁移。
订书肽策略也很快应用到多肽导向分子的设计上。RGD序列在脑部肿瘤递药领域被广泛研究,其整合素受体同时存在于新生血管和肿瘤细胞,具有促穿透和靶向的双重功能,但部分小鼠大脑内皮细胞的整合素受体表达并不高,胶质瘤早期完整的BBB对递药仍造成限制。为了进一步提高导向分子对BBB的穿透能力,同时维持其靶向肿瘤细胞的能力,Ruan等[36]设计了订书肽型的环状RGD(sRGD),在线性RGD序列的两端分别引入R8、S5交联氨基酸并利用烯烃复分解反应成环,提高其穿透屏障的能力。将sRGD修饰在载紫杉醇的PEG-PLA胶束上后,能够有效提高药物对BBB的穿透能力,同时维持了后续介导药物靶向脑肿瘤的能力。在小鼠U87脑胶质瘤模型上,基于订书肽策略设计的sRGD修饰的载紫杉醇胶束与普通环肽cRGD修饰相比,将荷瘤小鼠半数生存期从29 d延长至39.5 d。
2.2 免疫原性优化
传统认为,潜在免疫原性低是多肽较之大分子作为导向分子的可能优势之一。然而新近以来研究发现,当部分多肽导向分子与具有长循环功能的递药系统相结合后,其表现出较为明显的免疫原性,这关乎整个递药系统在体内的靶向效率以及安全性,今后需要重点关注。Wang等[37]发现经典的多肽导向分子c(RGDyK)修饰的脂质体在体内具有较强的免疫反应,甚至可诱发机体过敏性死亡。同时,环化和脂质体修饰均明显增强了c(RGDyK)最终的免疫原性,因此Wang等认为导向分子的构象和物理性质均可能对其免疫原性有较大影响。
Guan等[38]发现其前期设计的脑靶向多肽导向分子DCDX具有一定正电荷且肽链相对较长,导致修饰于脂质体表面后表现出一定的免疫原性,能与IgM发生吸附引起机体免疫反应并快速排出体外。为此,作者通过计算机辅助设计对DCDX进行优化,将其氨基酸序列大幅缩减到8个氨基酸并尽量去掉了带正电荷的氨基酸,所得D8多肽吸附IgM的量明显下降,有效降低了制剂在体内的免疫原性,增加其靶向效率。该工作表明,多肽配体修饰的脂质体,其表面蛋白冠吸附与配体的稳定性、电荷、大小、疏水性等性质密切相关,应尽量设计正电荷少、链短的多肽导向分子以调节表面蛋白冠吸附,最终实现对其免疫原性的优化。
2.3 导向分子的功能集成
经过筛选的多肽导向分子往往具有较高的亲和力,然而部分复杂的疾病往往会对导向分子的功能提出更多要求。除了对单一靶标的亲和力之外,在不同疾病的治疗中,还可能要求导向分子同时具有多种不同的性质,例如能协助跨生理屏障(如BBB、口服吸收屏障等)并靶向病灶、对多个不同靶标均具有靶向性等。为同时满足上述多种需求,有许多研究使用了多个功能不同的导向分子平行修饰,共同作用,然而进行复杂的多重修饰将对成药性造成不利影响,这提示多功能集成是多肽导向分子的发展需求之一。
iRGD(CRGDKGPDC)是由9个氨基酸构成的环肽,该片段含有能特异性结合整合素的RGD片段。与经典RGD相同,iRGD能与肿瘤新生血管内皮细胞上高表达的整合素受体αvβ3和αvβ5特异性结合,促进药物对该屏障的穿透。在结合整合素受体后,iRGD还能被酶切割激活,激活后该片段能与神经纤维毛蛋白受体1和2(neuropilin-1,neuropilin-2)受体结合,激活CendR通路,促进肿瘤细胞对药物的摄取[39]。因此,iRGD作为导向分子不需多重修饰,即可同时达到促血管穿透并靶向肿瘤的多重功能,故受到广泛关注。Liu等[40]将iRGD修饰在聚乳酸-羟基乙酸共聚物/壳聚糖纳米粒上,对卡莫司汀及其敏化剂进行递送,能够有效促进肿瘤细胞对药物的摄取,并在小鼠模型上实现了对BBB更强的穿透能力,以及在脑肿瘤部位更多的富集,取得了较好的治疗效果。
受到iRGD功能集成的启示,研究者们针对特定疾病需求,在针对单一靶点进行设计的基础上,对多个多肽或微型蛋白进行了序列拼接,或直接进行多功能的从头设计,最终达到在一个导向分子上按需进行功能集成的优化目的。例如,递送药物到病灶的过程中,可能需要克服多重生理屏障,要求导向分子集成跨不同生理屏障的能力,Belhadj等[41]针对脑部肿瘤递送中的BBB和血-脑肿瘤屏障,将RGD环肽和对羟基苯甲酸偶联为一个Y字型的集成导向分子,该导向分子既能发挥RGD环肽识别血-脑肿瘤屏障和胶质瘤的功能,又能发挥对羟基苯甲酸穿透BBB的能力。例如,将针对病灶不同靶点的导向分子进行集成后,相对单靶向的导向分子,其对病灶常具有更高的敏感性,尤其是在病灶具有异质性的情况下,其优势更为明显。胃泌素释放肽受体(gastrin-releasing peptide receptor, GRPR)和神经肽Y1受体(neuropeptide Y1 receptor,Y1)在乳腺癌细胞和前列腺癌细胞膜上都会过表达,为进一步提高导向分子对肿瘤的靶向能力,研究者们据此设计了若干集成的多肽配体。Ghosh等[42]将GRPR靶向配体和Y1靶向配体相拼接,设计了一种可实现对于GRP和Y1受体双靶向的多肽导向分子探针t-BBN/BVD15-DO3A,并在T-47D和MCF-7这2种乳腺癌细胞模型上证明了该集成导向分子的亲和力。随后,该课题组对该集成导向分子的血清稳定性和体外代谢降解情况进行了研究,通过研究发现双靶向多肽分子在血清中稳定性显著增加,具有进一步应用的潜力[43]。
3 发展趋势与展望
上述多肽导向分子的来源及优化部分实际上已涉及到大量的应用,因此本部分仅对多肽导向分子的应用趋势进行简述。随着相关领域的快速发展,多肽导向分子的应用已不再仅限于靶向病灶细胞本身。以多肽导向分子应用较多的肿瘤为例,导向分子的靶点一方面从单纯的靶向病灶延伸到递送过程中跨越各种脏器、组织乃至胞内的生理屏障,另一方面,目标也从肿瘤细胞延伸到更加复杂的肿瘤相关微环境,包括肿瘤新生淋巴管、肿瘤相关巨噬细胞甚至病理pH条件等[44-46]。
当然,多肽导向分子针对的疾病种类也从肿瘤延伸到了更大范围。例如,在匮乏新抗生素类药物的背景下,通过导向分子介导药物靶向病原微生物应该是和开发全新的小分子实体药物具有同等重要性。一些具有一定抗菌能力的抗菌肽同时也能扮演导向分子的功能,多肽-抗生素偶联物也开始引起关注[47-49]。而通过多肽文库同样也能筛选出针对具体病原微生物的识别分子,从而有效介导靶向治疗[50]。
尽管本身不是发挥药效的主要部分,导向分子的重要性并不亚于药效部分。针对新药研究的新技术和新策略同样可用于对多肽导向分子的性能优化。为平衡复杂疾病治疗需求和设计从简的成药性考量,功能集成的多肽导向分子将是今后发展和值得深入研究的重要方向。例如,针对不同靶点的多功能导向分子,其不同功能片段之间的比例、距离、连接段的刚性、对不同靶点的结合与解离动力学,以及结合后引发的一系列生理变化,甚至部分目标靶点之间的相互作用等,这些作用时机、作用动力学、生理功能等许多方面的问题,都是今后研究中值得考虑的因素。
疾病相关的靶受体大多具有内源性配体,有的甚至还存在内源性抗体。目前,多肽导向分子和上述内源性成分在相同靶受体上的识别位点往往是重叠的,这就可能会导致内源性的干扰或抑制。随着多肽蛋白相关生物学、化学及计算科学等的不断强力融合,针对目标蛋白更加“随心所欲”地获取多肽配体已变得可能[26]。通过避开靶受体的正位来设计能识别其“别位”的多肽配体,有望实现对内源性竞争的高效规避,从而在精准医学领域发挥更大的作用[51-52]。
猜你喜欢
天津农学院学报(2022年2期)2022-08-05
渤海大学学报(自然科学版)(2022年1期)2022-07-25
今日农业(2022年4期)2022-06-01
云南大学学报(自然科学版)(2022年3期)2022-05-25
猪业科学(2021年3期)2021-05-21
中华诗词(2019年1期)2019-11-14
分析化学(2017年12期)2017-12-25
科技创新导报(2016年30期)2017-03-15
环球人物(2015年12期)2015-09-10
科学中国人(2015年16期)2015-02-28
