
计算机辅助解析甲氨蝶呤/聚乙烯亚胺纳米粒组装机制与内在结构
2020-04-20 10:39雷远正孙丹云汤悦
中国医药导报 2020年7期
雷远正 孙丹云 汤悦
[摘要] 目的 纳米粒的组装机制与内部结构的表征是纳米粒研究领域中的难点,本文拟通过计算机模拟技术分析甲氨蝶呤/聚乙烯亚胺纳米组装体的组装机制与内在结构。 方法 通过分子对接、混合能计算与耗散动力学模拟等计算机模拟技术计算甲氨蝶呤/聚乙烯亚胺纳米组装体混合体系中的分子间作用力、混合能和介观模拟结构。结果 甲氨蝶呤与聚乙烯亚胺之间的分子间作用力为-12.98 kcal/mol,主要由静电作用、氢键作用和疏水作用组成;经过200 ns的耗散动力学模拟,甲氨蝶呤与聚乙烯亚胺形成了核壳结构的纳米聚集体。 结论 甲氨蝶呤/聚乙烯亚胺纳米组装体是以甲氨蝶呤分子为内核,聚乙烯亚胺分子为外壳的核壳结构纳米粒,而其组装的驱动力则主要由甲氨蝶呤的两个羧基与聚乙烯亚胺的氨基之间的强静电作用力提供。
[关键词] 纳米粒;分子对接;介观模拟;甲氨蝶呤
[中图分类号] R944 [文献标识码] A [文章编号] 1673-7210(2020)03(a)-0008-04
[Abstract] Objective The assembly mechanism and characterization of the internal structure of nano-drug are difficult problems in the field of nano-drug research. In this paper, the assembly mechanism and internal structure of Methotrexate/Polyethyleneimine nano-assembly were analyzed by computer simulation technology. Methods Computer simulation techniques such as molecular docking, mixing energy calculation and dissipation dynamics simulation were used to calculate intermolecular forces, mixing energy and mesoscopic simulation structures in the mixture system of Methotrexate/Polyethylenimine nanocomposites. Results The intermolecular force between methotrexate and polyethyleneimine was -12.98 kcal/mol, mainly composed of electrostatic action, hydrogen bond action and hydrophobic action. After 200 ns of dissipation dynamics simulation, methotrexate and polyethyleneimine formed a core-shell structure of nano-aggregate. Conclusion Methotrexate/Polyethyleneimine nanoassembly is core-shell nanoparticles with methotrexate as core and polyethyleneimine as shell. The driving force of the assembly is mainly provided by the strong electrostatic interaction between the two carboxyl groups of methotrexate and the amino group of polyethyleneimine.
[Key words] Nanomedicine; Molecular docking; Mesoscopic simulation; Methotrexate
甲氨蝶呤(methotrexate,MTX)是臨床上常用的一种细胞周期特异性细胞毒性药物,可用于包括急性白血病等多种恶性肿瘤的治疗[1-3]。传统的MTX口服片,由于MTX与消化道上皮细胞的长时间接触,极易引起上皮细胞的增殖抑制,介导胃炎、消化道出血等严重的胃肠副作用[4]。本课题组前期通过聚乙烯亚胺(polyethyleneimine,PEI)与MTX的自组装成功构建了超高载药性能的MTX口服纳米制剂,但其组装机制仍有待进一步探索[5]。
分子间相互作用力是两个分子特异性非共价键识别的关键[6-7]。在分子识别过程中,空间立构、静电作用及疏水作用发挥着重要作用[8]。随着计算化学、计算机科学与药学等交叉学科的快速发展,计算机辅助技术已被广泛的应用于新药研发的药物筛选、药物设计与药理作用机制的分析中[9-12]。本课题组通过计算机模拟已成功实现了对纳米组装机制、载药机制、响应机制与药物释放机制的模拟[13-15]。受此启发,本研究通过Autodock 4.2软件对PEI与MTX之间的分子间作用力进行分析[16],并通过耗散粒子动力学(dissipative particle dynamics,DPD)[17-20]模拟分析PEI与MTX组装过程,从而解析MTX/PEI纳米粒的组装机制。
1 资料与方法
1.1 3D空间构象的准备与优化
在Drugbank数据库中下载MTX的分子结构式,在Matreilas Studio 2016软件中打开,加氢后,采用Forcite模块中的Geometry Optimization任务对MTX的3D结构进行能量最小化处理,获得合理的MTX的3D空间构象。考虑到计算机的计算能力,在Materials Studio 2016中利用分子结构的编辑功能构建n = 20的PEI的分子结构式,并进一步采用Forcite模块中的Geometry Optimization任务对MTX的3D结构进行能量最小化处理。
1.2 Autodock分子对接
在AutoDockTools 4.2中以受体导入PEI的分子结构,进行加氢和加电荷后以PDBQT格式存储。随后以配体导入MTX的分子结构,添加电荷后以PDBQT格式存储。在准备好配体与受体分子构象后,使用AutoGrid进行能量格点计算,Gird Box大小设置为126×126×126个网格点,每个小网格点的距离为0.0375 nm。采用拉马克遗传算法(lamarckian,GA)进行构象搜索,初始种群数设置为100,能量评定最大次数为2 500 000,其他参数采用默认值进行分子对接计算。
1.3 分子间混合能的计算
将优化后的水分子、PEI和MTX分子导入到Materials Studio 2016的工作界面,采用Blends模块中的Mixing任务,在Compass 2.0力场下进行混合能的模拟计算,模拟质量选择Fine,其他参数默认。
1.4 DPD介观模拟
1.4.1 PEI、MTX与水的粗粒化处理 将PEI、MTX与水分子分别设置为3个DPD粒子,分别命名为P、M和W,粒子半径设置为6.46 ?魡,分子量设置为54。其中PEI的粒子对应着20个重复的PEI单元,MTX的粒子MTX对应着1个MTX分子,而水分子的粒子Water对应着3个水分子。
1.4.2 珠子间相互作用参数的计算 取密度ρ = 3,则珠子间相互作用参数aij与Flory-Huggins相互作用参数(χ)之间存在以下线性关系:
基于Blends中计算的Flory-Huggins参数,最终计算出P、M与W珠子间的相互作用参数,并进一步构建DPD力场,用于后续的DPD模型的构建与模拟。
1.4.3 DPD模型的构建与优化 首先,基于PEI、MTX与水分子的珠子,构建PEI、MTX与Water的介观分子,随后按照1∶1∶8的比例构建PEI、MTX与Water的混合模型,其中Length scale项设为6.46 ?魡,Mass Scale项设为54 Amu,Density项设置为3 g/cm3,点击Build,就可得到水分子和MTX分子和PEI分子的混合分子的介观模型。并进一步选用构建的DPD力场,对所构建的介观模型进行优化。得到构象优化后的介观模型后,基于DPD力场,进一步采用Mesocite模块进行200 ns的耗散动力学模拟。
2 结果
2.1 PEI、MTX与水分子之间的分子间作用力分析
采用Autodock 4.2软件,计算PEI、MTX与水分子之间的结合能、分子间作用力、范德华力与静电作用力。从表1中可见,MTX与PEI之间的分子间作用力最大,为-12.98 kcal/mol,其中静电作用力贡献最大,达到了-11.36 kcal/mol,因此其结合到PEI上的结合能也远大于PEI和水分子结合到PEI上的结合能,达到了-10 kcal/mol。见表1。
进一步对MTX结合至PEI的最低能量构象进行分析。MTX与PEI之间主要通过MTX中的羧基与PEI中的氨基形成静电作用和氢键作用。见图1。
2.2 PEI、MTX与水分子之间分子间混合能的计算结果
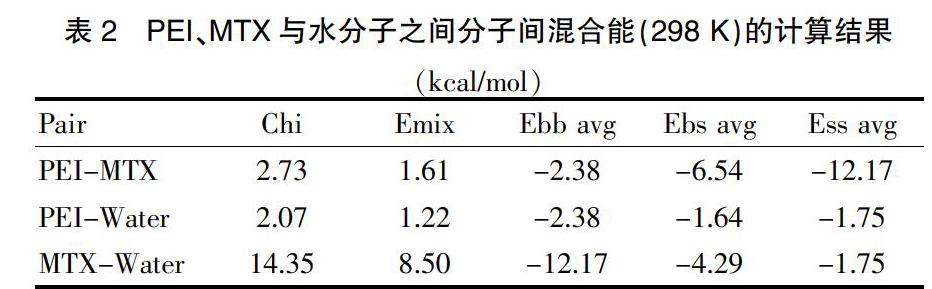
经计算,PEI与MTX之间的混合能为1.61 kcal/mol,远低于MTX与水分子之间8.50 kcal/mol的混合能大小。见表2。
2.3 组装体的介观模拟结果
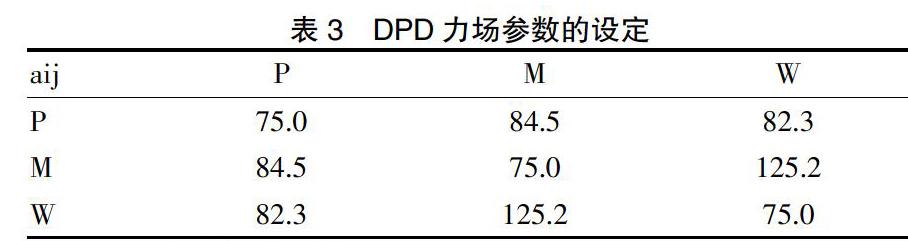
通过Blends模块计算的Flory-Huggins参数(Chi),将PEI(n = 20)、MTX和3个水分子分别设定为P、M和W等3個珠子,并通过表1中Flory-Huggins参数,进一步计算得到珠子间的DPD排斥参数aij。见表3。随后,基于珠子,分别构建了分子量为20 000的PEI的介观分子(约25个珠子)模型,与MTX和Water的介观分子模型。
进一步以PEI、MTX和水分子1∶1∶8的比例构建了水、MTX和PEI的混合的介观体系,并基于设定的DPD力场,对其构象优化,获得了其初始介观体系。见图2(封三)。
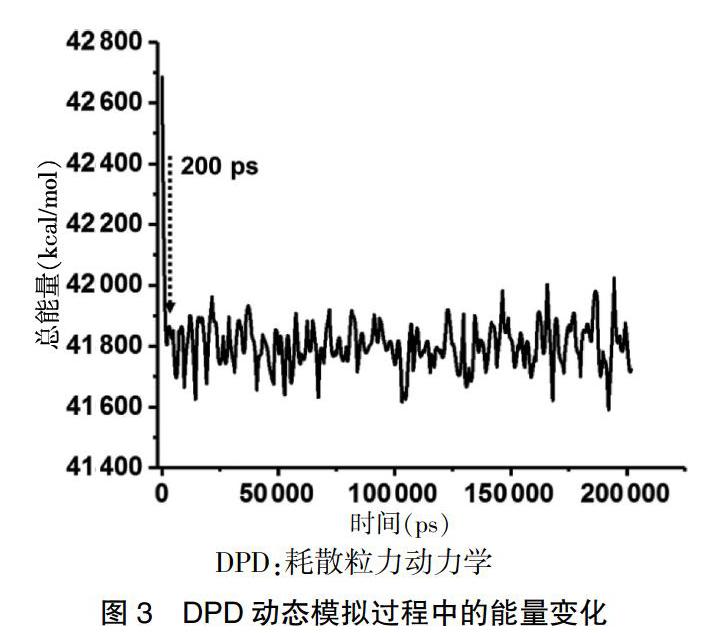
在得到初始介观模型后,基于设定的DPD力场,进一步采用Mesocite中的DPD模块进行了200 ns的DPD动态模拟。通过对DPD动态模拟过程中体系总能量变化的跟踪。见图3,可以发现体系的总能量在200 ps即发生了锐减,随后就稳定在41 800 kcal/mol上下,提示200 ns的模拟时间足够得到稳定的介观组装结构。
经过200 ns的分子模拟过程,最终获得了一个球形的组装体形态,其表面由桔色的PEI介观分子呈网络状覆盖。见图4(封三)。
通过设置横截面,可以清晰观察到该介观组装体的内部结构。见图5(封三)。由大量的MTX介观分子填充形成内核,而其外围由PEI介观分子覆盖形成外壳。
3 讨论
本研究通过对MTX、PEI和水分子混合体系中分子间作用力、混合能以及DPD介观模拟的计算与分析,成功揭露了MTX/PEI纳米组装体的组装机制与内部结构。AutoDock分子对接结果提示,MTX与PEI之间较强的分子间作用力,特别是静电作用力,是支持MTX/PEI纳米组装体组装成功的原始动力。混合能的计算结果显示,在水分子体系中,MTX与聚乙烯亚胺之间的混合相容性要优于于MTX与水分子之间的混合相容性。因此,在MTX、水分子和PEI的混合体系中,MTX分子更倾向于与PEI混合,而倾向于避免与水分子的接触。随后的DPD介观模拟结果显示,MTX/PEI纳米组装体是以MTX分子为内核,PEI分子为外壳的核壳结构纳米粒。
随着计算机技术的迅速发展,研究者基于计算机模拟技术已经大幅提高了对于生物大分子结构与功能的理解,但将计算机模拟技术应用于药剂学领域的研究尚处于萌芽阶段。特别是纳米粒极小的纳米尺寸导致其难以被充分表征,提高了通过传统的实验表征手段揭露其组装机制与内部结构的难度。本研究创新性的将计算机分子模拟技术引入到纳米粒的组装机制与内部结构的表征中,实现了对PEI/MTX纳米组装体组装机制与内部结构的分子模拟。
[参考文献]
[1] Chan ES,Cronstein BN. Methotrexate—how does it really work? [J]. Nat Rev Rheumatol,2010,6(3):175-178.
[2] Chan ES,Cronstein BN. Mechanisms of action of methotrexate [J]. Bull Hosp Jt Dis(2013),2013,71 Suppl 1:S5-S8.
[3] Tian H,Cronstein BN. Understanding the mechanisms of action of methotrexate:implications for the treatment of rheumatoid arthritis [J]. Bull NYU Hosp Jt Dis,2007,65(3):168-173.
[4] Gutierrez JC,Hwang K. The toxicity of methotrexate in male fertility and paternal teratogenicity [J]. Expert Opin Drug Metab Toxicol,2017,13(1):51-58.
[5] 蒙小丽,龙玲,陈小勇.基于聚乙烯亚胺构建pH响应性口服甲氨蝶呤纳米递送系统[J].中国当代医药,2018,25(4):20-24.
[6] 陆小华,朱育丹,邬新兵,等.基于分子间相互作用的纳米尺度分子传递[C]//第十七届全国化学热力学和热分析学术会议论文集,2014.
[7] 徐筱杰,陳丽蓉.化学及生物体系中的分子识别[J].化学进展,1996,8(3):189.
[8] Náray-Szabó G. Analysis of molecular recognition:steric electrostatic and hydrophobic complementarity [J]. J Mol Recognit,2010,6(4):205-210.
[9] Kapetanovic IM. Computer-aided drug discovery and development(caddd):in silico-chemico-biological approach [J]. Chem Biol Interact,2008,171(2):165-176.
[10] Talele TT,Khedkar SA,Rigby AC. Successful applications of computer aided drug discovery:moving drugs from concept to the clinic [J]. Curr Top Med Chem,2010,10(1):127-141.
[11] Yang X,Wang Y,Byrne R,et al. Concepts of Artificial Intelligence for Computer-Assisted Drug Discovery [J]. Chem Rev,2019,119(18):10520-10594.
[12] Yang B,Mao J,Gao B,et al. Computer-Assisted Drug Virtual Screening Based on the Natural Product Databases [J]. Curr Pharm Biotechno,2019,20(4):293-301.
[13] Zhang C,Long L,Xiong Y,et al. Facile Engineering of Indomethacin-Induced Paclitaxel Nanocrystal Aggregates as Carrier-Free Nanomedicine with Improved Synergetic Antitumor Activity [J]. ACS Appl Mater Inter,2019,11(10):9872-9883.
[14] Zhou X,Zhao Y,Chen S,et al. Self-Assembly of pH-Responsive Microspheres for Intestinal Delivery of Diverse Lipophilic Therapeutics [J]. Biomacromolecules,2016, 17(8):2540.
[15] Che L,Liu Z,Wang D,et al. Computer-assisted engineering of programmed drug releasing multilayer nan-omedicine via indomethacin-mediated ternary complex for therapy against a multidrug resistant tumor [J]. Acta Biomater,2019,97:461-473.
[16] Morris GM,Ruth H,William L,et al. AutoDock4 and AutoDockTools4:Automated docking with selective receptor flexibility [J]. J Comput Chem,2010,30(16):2785-2791.
[17] Warren PB. Dissipative particle dynamics [J]. Curr Opin Colloid In,1998,3(6):620-624.
[18] Hoogerbrugge PJ,Koelman JMVA. Simulating Microscopic Hydrodynamic Phenomena with Dissipative Particle Dynamics [J]. Europhys Lett,1992,19(3):155-160.
[19] Espanol P,Warren P. Statistical mechanics of dissipative particle dynamics [J]. Europhys Lett,1995,30(4):191-196.
[20] Moyassari A,Gkourmpis T,Mikael S,et al. Molecular dynamics simulation of linear polyethylene blends:Effect of molar mass bimodality on topological characteristics and mechanical behavior [J]. Polymer,2019,161:139-150.
(收稿日期:2019-08-12 本文编辑:封 华)
猜你喜欢
现代临床医学(2021年2期)2021-03-29
中华养生保健(2020年9期)2021-01-18
风湿病与关节炎(2017年3期)2017-04-05
中国中药杂志(2017年6期)2017-04-05
中国中药杂志(2017年4期)2017-03-28
中国中药杂志(2017年3期)2017-03-20
科技创新与应用(2016年34期)2016-12-23
江苏农业科学(2015年5期)2015-10-20
中国当代医药(2015年30期)2015-03-01
中国当代医药(2015年9期)2015-03-01
