
Li修饰的石墨炔纳米管储氢性能的第一性原理研究
2020-07-10 01:13陈泽新白忠臣秦水介
四川大学学报(自然科学版) 2020年4期
陈泽新, 白忠臣, 秦水介
(贵州大学 大数据与信息工程学院, 贵阳550025; 贵州大学 贵州省光电子技术及应用重点实验室, 贵阳550025)
1 引 言
人类对能源的追求就从未停止, 氢因拥有高燃烧值、可再生、环保等优点被人们格外关注. 氢的储运是限制氢能源使用的关键性问题. 碳纳米材料因其具有良好的可逆性、吸放氢条件温和等优秀条件, 被赞誉为富有储氢应用前景值得开发的材料[1-3]. 但单一的纳米材料与氢分子的结合能较弱, 储氢量较低[4]. 2005年美国再生能源国家实验室用过渡金属修饰C60和C48B12后发现其储氢量大大提高[5], 但后来进一步研究发现过渡金属在C60表面吸附不稳定会形成团簇将大大降低储氢量.为了解决这个问题, 近年来开始关注石墨炔这种在光学和电子学方面有新奇特性的材料. 石墨炔和石墨烯具有同样的对称性, 能看作是将石墨烯中的三分之一的C-C键用C=C键取而代之形成的一种类石墨烯的单层结构[6-7], 由于石墨炔表面的孔洞, 金属在表面吸附不会发生聚合现象. 2011年Li等[8]研究表明Ca修饰的石墨炔储氢的质量比为9.66 wt%, 吸附能在0.2 eV/H2左右, 吸附性能比较良好是因为石墨炔拥有π键.
石墨炔片层材料拥有良好的吸附性能, 因此探究它的管状立体式吸附结构是否会有更好地特性. 石墨炔纳米管结构[9]可看作是石墨炔卷成的, 研究表明γ型石墨炔是比较稳定的结构, 它形成的γ型石墨炔纳米管也是比较稳定的[10]. 2013年王玉生[11]研究了Ca修饰的石墨炔纳米管, 吸附能在0.13~0.33 eV/H2之间, 每个Ca原子可以吸附4个氢分子. 由于Ca原子相对质量比较大, 储氢质量比不高, 故尝试另外一种过渡金属进行修饰. 很多研究发现Li修饰的材料拥有良好的储氢性能[12-13], 它的原子质量较低, 因此运用基于第一性原理密度泛函理论(DFT)的计算方法研究了Li修饰的γ型石墨炔纳米管的储氢性能, 模拟分析其在储氢领域的性能并探寻其应用潜力. 这些研究可为今后设计高性能的储氢纳米材料奠定理论基础.
2 理论模型与计算方法
本文应用基于第一性原理密度泛函理论(DFT)的第一性原理计算方法(CASTEP软件包)对石墨炔模型进行构建和计算. 石墨炔是由苯环链接炔键共轭形成的, 结构是全碳分子的二维平面网状, 它的晶格参数为a=1.407 Å,b=1.394 Å,c=1.226 Å, 空间夹角分别是α=90°,β=90°,γ=120°. 由于γ型石墨炔纳米管是最稳定的, 因此选用γ相单胞作为基础模型进行卷管得到管径为zigzag型(2,1)的单胞模型. 本研究对石墨炔纳米管单胞布里渊区的K点选取为2×2×1, 最佳截断能为300 eV, 能量收敛精度1.0×10-5eV/atom, 原子间相互作用力收敛标准为0.01 eV/Å, 最大应力偏差精度设置为0.05 GPa.
3 计算分析与讨论
3.1 Li修饰石墨炔纳米管的稳定性及性能
石墨炔片层理论模型有三种, 如图1(a)是α-graphdyne片层理论模型, 碳碳单键、双键以及三键键长分别为1.388、1.240和1.410 Å. 如图1(b)是β-graphdyne片层理论模型, 碳碳单键、双键以及三键键长分别为1.43、1.340和1.2Å. 如图1(c)是γ-graphdyne片层理论模型, 碳碳单键、π键以及三键键长分别为1.407、1.394和1.226 Å.
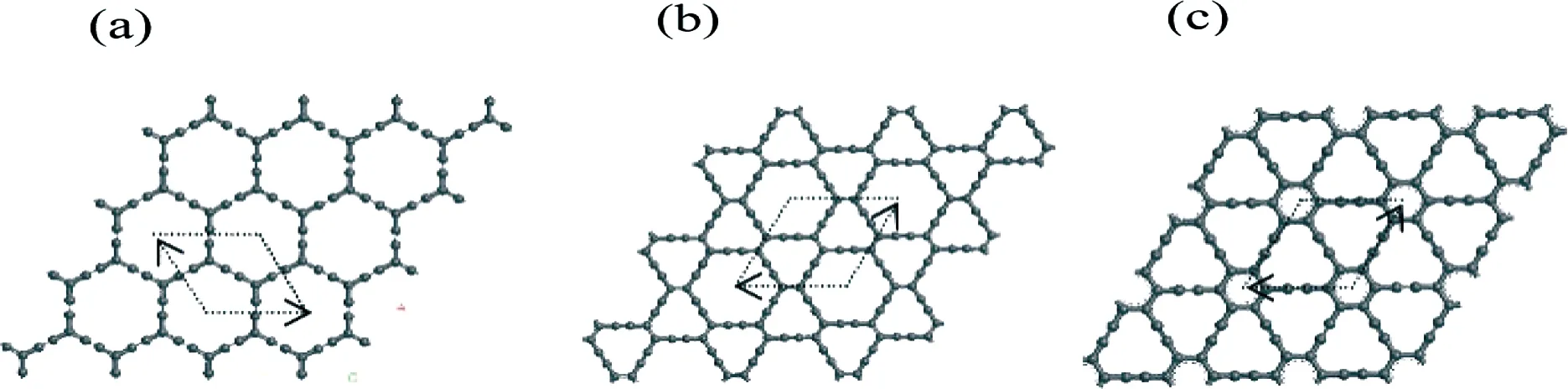
图1 (a) α石墨炔片层模型; (b) β石墨炔片层模型; (c) γ石墨炔片层模型Fig.1 (a) α-graphdyne; (b) β-graphdyne; (c) γ-graphdyne
分别对石墨炔α、β、γ相片层模型进行优化与计算得到γ相体系的能量最低的结果, 因此γ相石墨炔是三种结构中稳定性最好的. 将γ相石墨炔二维片层理论模型沿着n方向的片层模型卷曲, 石墨炔纳米管沿着m方向生长, 构建的为zigzag型γ-GNT. 沿着m方向将石墨炔片层模型卷曲,石墨炔纳米管沿着n方向生长, 构建的为armchair型γ-GNT. 本文选择zigzag型γ-GNT作为研究对象, 图2是管径为zigzag(2,1)γ-GNT理论模型,α=β=90°,γ=120°.
本文计算了(2,1)-(12,6)管径的石墨炔纳米管的能带结构如图2. 从图2中可以看出石墨炔纳米管的导带底和价带顶位于同一位置, 证明了石墨炔纳米管属于直接带隙半导体材料, 并且研究不同管径得出的禁带平均宽度在0.38~0.63 eV范围与参考文献中所述一致[14].
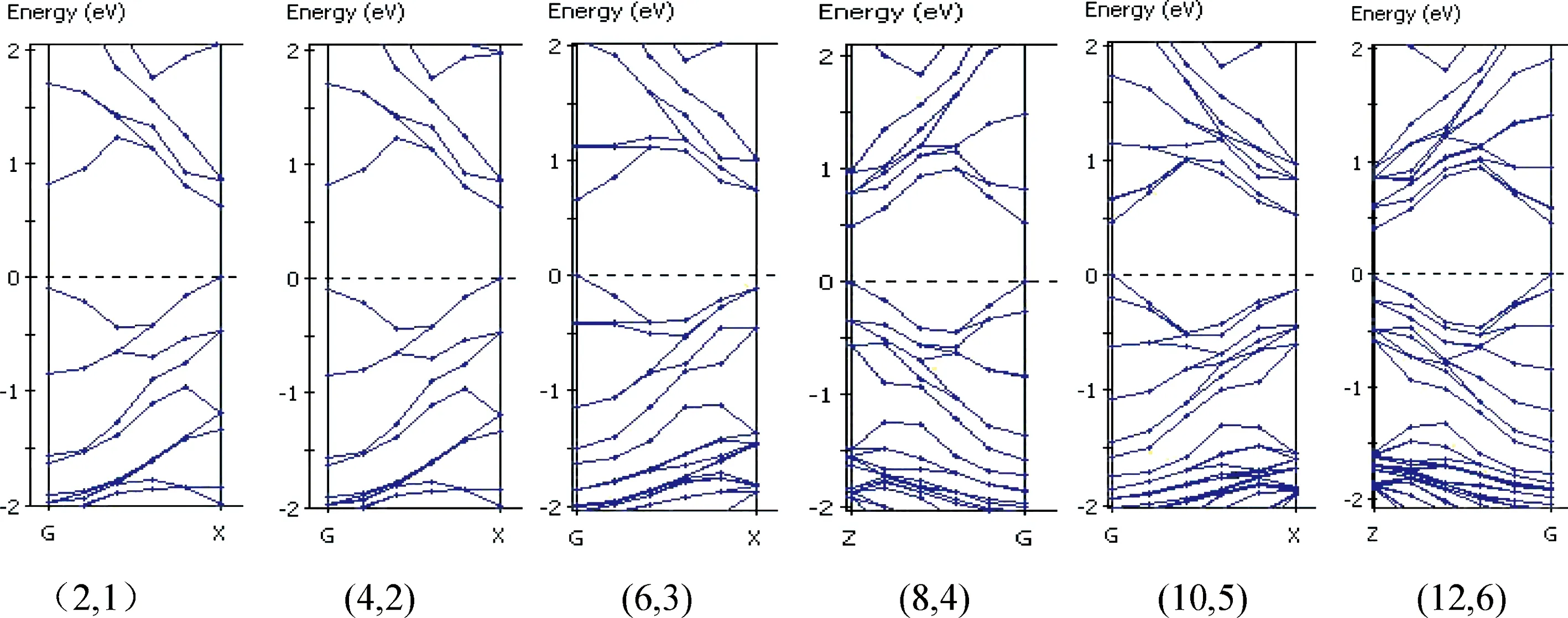
图2 zigzag型γ-石墨炔管(2,1)~(12,6)能带结构图Fig.2 Band structures of zigzag (2,1)~(12,6)-GNT
为了进一步研究石墨炔纳米管被修饰后的性能, 选择建立管径为zigzag型(2,1)γ相石墨炔管, 该体系包含48个C原子, 如图3.

图3 zigzag型γ-GNT理论模型Fig.3 Theoretical structure model of zigzag γ-GNT
研究表明氢的有效吸附能选择在0.13~0.7 eV[11]之间, 吸附能太小则无法吸附氢, 吸附能太大则不利于氢的利用. 定义Li修饰的石墨炔纳米管储存氢的吸附能公式为[15]:
(1)
其中Ead表示吸附能,n表示H2分子的数目,E(Li/GNT)、E(H2)、E[n(H2)+Li/GNT]分别表示Li修饰GNT后体系的总能量、一个孤立的H2分子的能量、Li修饰的GNT体系吸附n个H2分子后体系的总能量.
定义被Li修饰的石墨炔纳米管计算总能量的公式为:
Eb=E(GNT)+E(Li)-E(GNT+Li)
(2)
其中Eb表示总能量,E(GNT)代表石墨炔纳米管体系能量,E(Li)表示Li原子能量,E(Li+GNT)表示Li修饰石墨炔纳米管后的体系能量.
为了使研究目标具有一致性和普遍性, 研究一个Li原子在zigzag型γ-GNT单胞模型上的吸附情况. zigzag型γ-GNT单胞模型上有五个位置提供给Li进行修饰, 分别是六元环的中心, 三个不同的C-C键的桥位, 乙炔键所围成的十二元环. 进行结构优化后, 根据公式(2)分析后表明Li原子吸附在乙炔键所围成的十二元环位置能量为-7.599 164 74×103eV, Li原子吸附在六元环位置的能量为-7.598 260 57×103eV, 而其他三个桥位不稳定, 经过弛豫Li原子会移动到十二元环处, 能量最低的位置为乙炔键所围成的十二元环处, 因此它是最佳修饰位, 如图4.
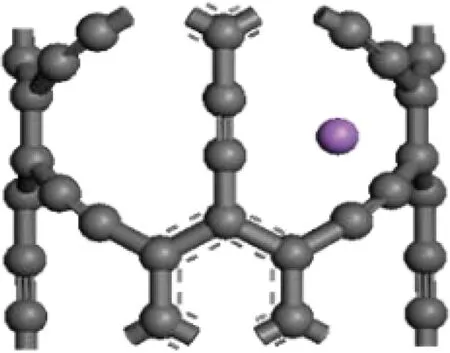
图4 Li原子处于十二元环修饰位
Fig.4 Structure of Li on the twelve bonds constituted by acetylene of GNTs
为了验证经过Li修饰的zigzag型γ-GNT单胞模型的储氢性能比没修饰过的优越, 给出了zigzag型γ-GNT单胞模型吸附氢, Li修饰的zigzag型γ-GNT单胞模型上未吸附氢, 以及Li修饰的zigzag型γ-GNT单胞模型吸附氢的分波态密度图, 如图5, 6, 7所示.
图5(a)与(b)是未修饰的zigzag型γ-GNT单胞模型吸附氢分子中氢原子与碳原子的分波态密度图, 从图5看出在费米能级处, H的s轨道只有微弱的能量, 与C的相互作用微弱是由于静电作用力为主要吸附力, 因此吸附氢的能力比较弱.
图6(a)与(b)是Li修饰zigzag型γ-GNT单胞模型未吸附氢分子中锂原子与碳原子的分波态密度图, 从图6中可看出在费米能级处, Li的s轨道与C的p轨道有轨道杂化[11], 因此两者之间有较强的作用力, 比较稳定.
从图7中可看出, GNT与Li有良好的轨道杂化, 多了化学键的作用, H也比纯净时的能量有所提高, Li在其中充当桥梁作用, 使石墨炔对氢的吸附能力变强. Li修饰的zigzag型γ-GNT单胞模型的稳定性较高, H与Li之间的轨道杂化和静电场作用以及体系的化学键作用力共同提高了整个体系的吸附能[11], 它能够储存更多的H2分子, 证明了用Li修饰石墨炔纳米管的理论可行性.
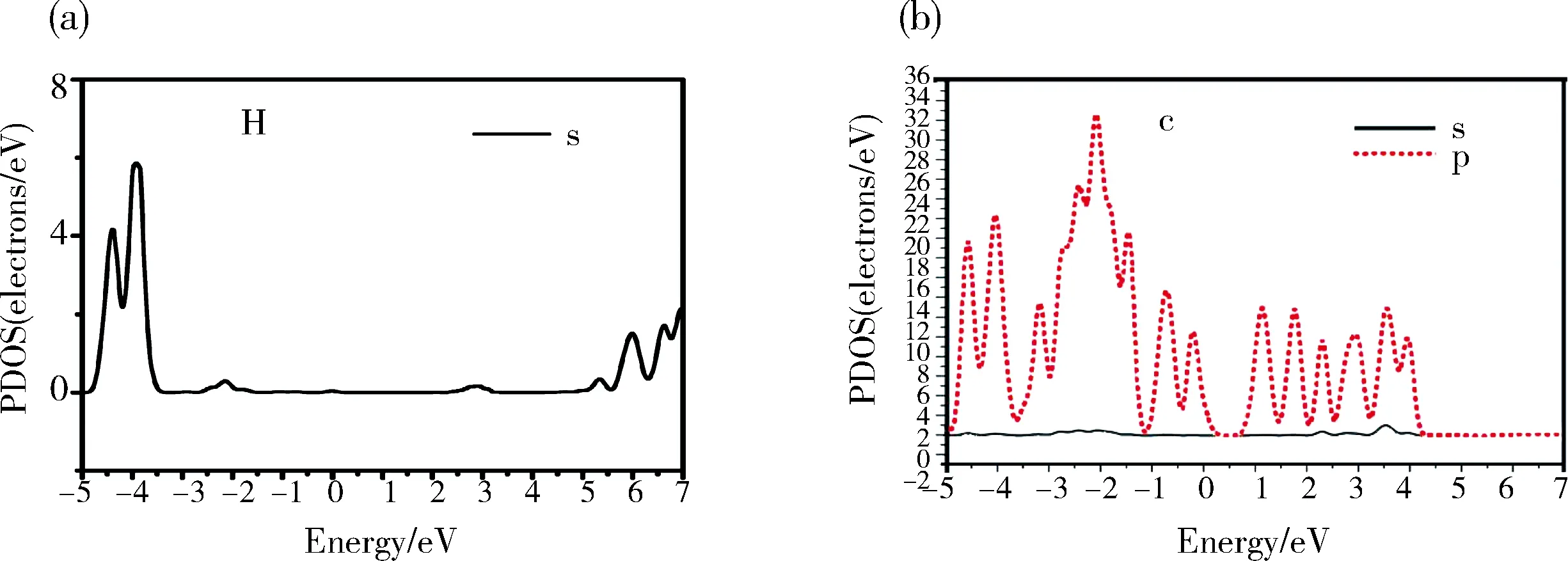
图5 (a)石墨炔纳米管吸附氢中H原子的分波态密度图;(b)石墨炔纳米管吸附氢中C原子的分波态密度图
Fig.5 (a) Partial density of states (PDOS) of H in GNTs adsorb hydrogen molecule; (b) PDOS of C in GNTs adsorb hydrogen molecule
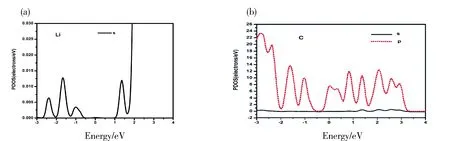
图6 (a)Li修饰石墨炔纳米管未吸附氢中Li原子的分波态密度图;(b)Li修饰石墨炔纳米管未吸附氢中C原子的分波态密度图
Fig.6 (a)PDOS of Li in Li decorated GNTs; (b) PDOS of C in Li decorated GNTs
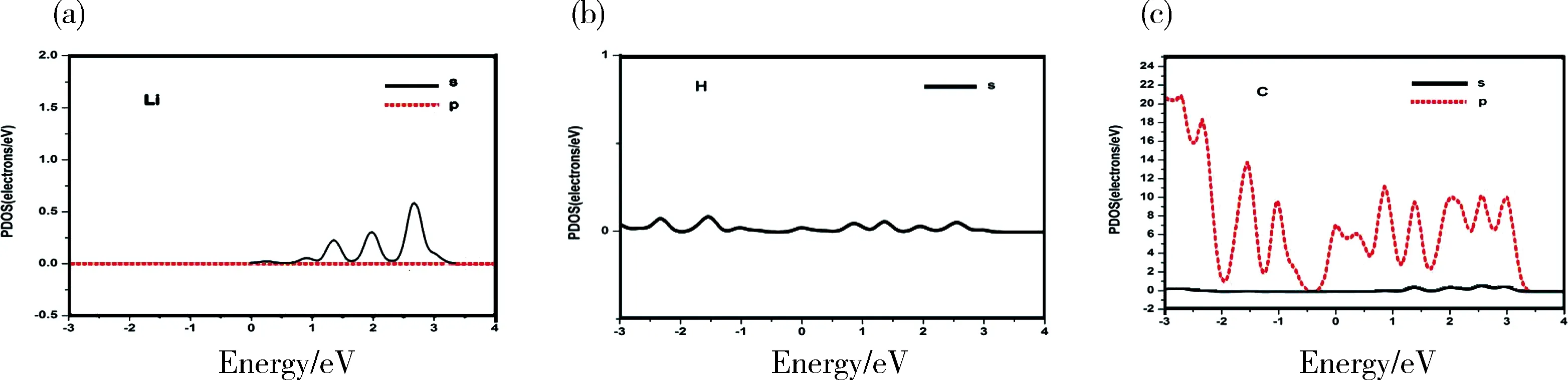
图7 (a) Li修饰的石墨炔纳米管吸附氢中Li原子的分波态密度图; (b) Li修饰的石墨炔纳米管吸附氢中氢原子的分波态密度图; (c) Li修饰的石墨炔纳米管吸附氢中C原子的分波态密度图
Fig.7 (a) PDOS of Li in Li decorated GNTs adsorb hydrogen molecule; (b) PDOS of H in Li decorated GNTs adsorb hydrogen molecule; (c) PDOS of C in Li decorated GNTs adsorb hydrogen molecule
3.2 储氢性能的研究
在找到Li修饰的zigzag型γ-GNT单胞模型最佳修饰位后, 开始往最佳修饰位处添加氢分子, 加入一个氢分子之后通过CASTAP软件包优化和计算得出Li修饰的石墨炔纳米管的能量, 通过公式(1)计算出氢的吸附能, 再加入第二个氢分子, 再算出氢的吸附能, 依次类推, 得到的分子模型如图8, 计算结果如表1.
从表1看出, 随着氢分子的加入系统的吸附能逐渐降低. 当Li修饰的石墨炔纳米管吸氢分子数达到8时, 它的吸附能为0.119 503 8 eV, 小于氢分子要求的最低吸附能0.13 eV. 因此系统无法
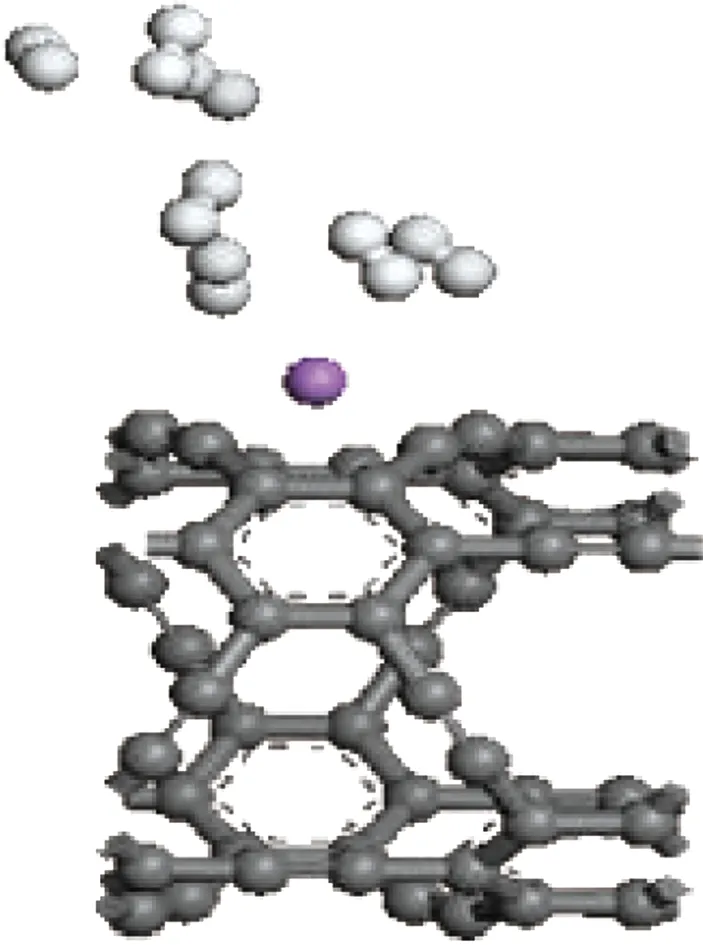
图8 Li修饰的石墨炔纳米管吸附七个氢分子模型
Fig.8 Structure of Li decorated GNTs adsorb 7 hydrogen molecules
吸附8个氢分子, 它的最大储氢量为7个氢分子. 只有1个Li修饰GNT时, 吸附的H2分子在该体系中的质量比达到2.34 wt%, 吸附能在0.13~0.36 eV/H2. 因此可以看出Li修饰的石墨炔纳米管在储氢性能方面具有极大的优势和潜力.
表1 锂修饰的石墨炔纳米管吸附能统计表
Tab.1 Adsorption energies of Li decorated graphdiyne nanotubes
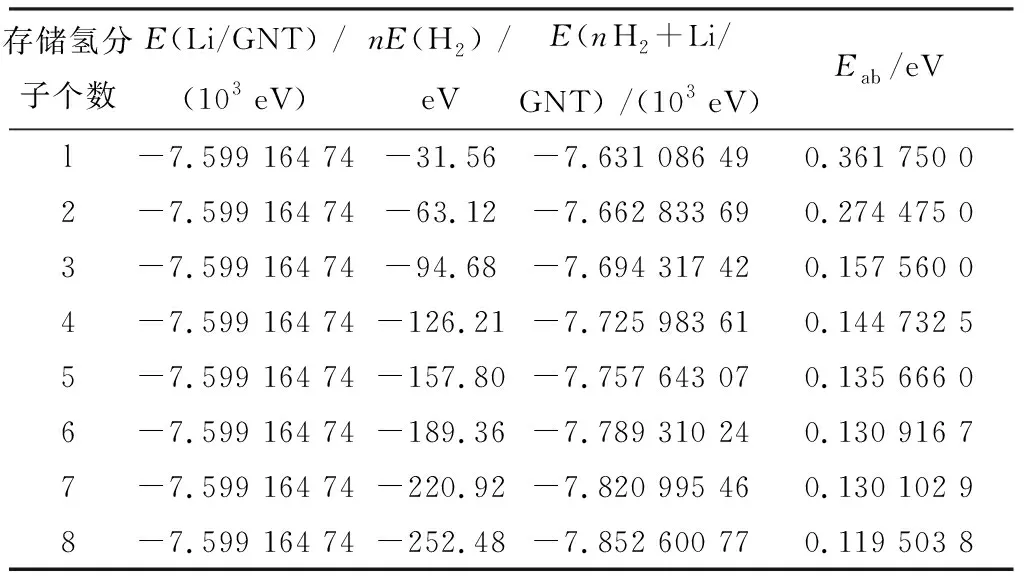
存储氢分子个数E(Li/GNT) /(103 eV)nE(H2) /eVE(nH2+Li/GNT) /(103 eV)Eab/eVl-7.599 164 74-31.56-7.631 086 490.361 750 02-7.599 164 74-63.12-7.662 833 690.274 475 03-7.599 164 74-94.68-7.694 317 420.157 560 04-7.599 164 74-126.21-7.725 983 610.144 732 55-7.599 164 74-157.80-7.757 643 070.135 666 06-7.599 164 74-189.36-7.789 310 240.130 916 77-7.599 164 74-220.92-7.820 995 460.130 102 98-7.599 164 74-252.48-7.852 600 770.119 503 8
4 结 论
通过基于密度泛函理论的第一性原理方法研究了Li修饰γ石墨炔纳米管的储氢性能得到了以下结论:(1) Li原子能稳定的吸附在zigzag型γ-GNT单胞模型上, 它的最佳修饰位是乙炔键所围成的十二元环处. (2) 随着Li修饰的zigzag型γ-GNT单胞模型吸附氢分子个数的增加, 氢的吸附能逐渐降低, 由于吸附氢分子数达到8时, 氢的吸附能小于0.13 eV, 因此系统在常温下最大吸附氢的个数为7个H2分子. (3) 单个Li修饰zigzag型γ-GNT单胞模型, 吸附的H2分子在该体系中的质量比达到2.34 wt%, 吸附能在0.13~0.36 eV/H2, 而以往的单个Ca修饰zigzag型γ-GNT单胞模型时只能吸附4个H2分子且质量比仅为1.28 wt%[11], 因此该系统吸附能是比较理想的并且性能优秀的.
综上所述, 本文研究的Li修饰的石墨炔纳米管储氢性能优越为设计储氢性能材料提供了理论基础.
猜你喜欢
人工晶体学报(2022年6期)2022-07-30
汽车实用技术(2022年10期)2022-06-09
人工晶体学报(2022年3期)2022-04-14
无机盐工业(2022年3期)2022-03-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
汽车实用技术(2018年7期)2018-05-18
分析化学(2018年12期)2018-01-22
汽车文摘(2016年10期)2016-12-07
