
耳畸形诊疗策略
2020-10-14 13:25苏钰
中国听力语言康复科学杂志 2020年3期
苏钰
耳的主要功能是感知声音。为了把听功能发挥到极致,人类在进化过程中,形成了外耳、中耳、内耳三个部分,协同进行声的收集、传递和转化,是精密的人体感觉器官。从耳的发育过程来看,外、中、内耳来自不同胚层,每一部分又以组合的方式形成,因而发育中的微小事件极易导致耳的形态和功能异常。约20%的新生儿出生后会存在耳廓变形,小耳畸形(外中耳畸形)发生率约0.01%~0.02%[1]。一般来讲,小耳畸形极少同时伴有内耳畸形,内耳畸形通常单独发生,约占先天性耳聋患者的20%[2]。
耳廓畸形发生率高但轻重程度不一,轻微的仅有耳廓变形不伴有组织缺损,严重的则可能存在耳廓组织的不同程度缺损、伴或不伴外耳道狭窄或闭锁,也可能合并半面短小、面瘫、神经性耳聋,甚至可能为某些涉及全身系统综合征的一部分[3]。
因为同时涉及功能及外观,且部分疾病类型具有宝贵治疗时间窗,治疗时需根据不同的畸形类型、患儿发育不同阶段进行专业的序贯性治疗。需要相关专科医生兼具耳外科、整形外科、颌面外科、听力学、影像学及遗传学等相关知识。因而,耳畸形治疗的多学科合作和个体化设计非常重要。
根据治疗方案的不同,耳畸形临床实用分类可分为:①外耳的形态畸形(无明显的耳廓组织缺损以耳廓的变形为主);②外耳结构畸形(伴有耳廓组织缺损及外耳道狭窄或闭锁,多合并中耳畸形,即小耳畸形,也称外中耳畸形);③单纯性中耳畸形;④内耳畸形;⑤综合征性耳畸形。本文就不同类型耳畸形的诊疗策略进行系统阐述。
1 耳畸形治疗方案的选择
1.1 外耳的形态畸形
耳廓形态畸形是指耳廓的变形,指耳廓没有明显的结构缺损,或缺损很轻。常见有收缩耳(又称杯状耳或垂耳)、招风耳、隐耳、Stahl's耳等,多不伴有外耳道狭窄或闭锁、一般不影响听力,可以在新生儿期(出生1个月内,最佳时间3~21天)采用耳矫形器进行无创矫治,对于一部分有轻微组织缺损的耳廓畸形,可以在新生儿期应用矫正器矫治,为将来手术做准备[4]。对于超出治疗时间窗的耳廓形态畸形及附耳(又称拴马桩)、耳垂裂等,可在患者6岁以后采用局部软骨及组织重塑的方法进行修整,但其中部分类型的耳廓变形后期手术效果不甚理想。国内家长普遍对耳廓形态畸形的了解、认知、重视程度不足,一般都持等待+观望的态度,寄希望于耳廓的自行矫正,甚至许多产科、儿科、耳鼻喉科及整形科医生也对该病的早期治疗没有足够的认识和重视。遇到家长拒绝矫治的情况,一般尊重家长的选择,但如果患儿存在耳道及耳甲腔软骨的变形导致外耳道狭窄,还是要积极建议家长接受治疗,避免后继外耳道狭窄引起听力障碍和外耳道胆脂瘤等并发症的出现。因有矫治最佳时间的限制,目前阶段亟需对耳形态畸形的非手术矫治相关知识进行普及和推广,参考梅奥诊所和笔者前期的工作经验,可与产科联合,同时将新生儿耳廓形态筛查与新生儿听力筛查结合起来,进行筛查人员规范化培训,使筛查窗口前移,及时诊断治疗[5,6]。
1.2 外耳结构畸形(伴有耳廓组织缺损、外耳道狭窄或闭锁及中耳畸形)
外耳结构畸形一般指小耳畸形,因多同时伴发中耳畸形,也叫外中耳畸形。小耳畸形是先天性耳廓畸形中比较严重的一型,常伴有外耳道狭窄或闭锁、中耳畸形、中耳胆脂瘤形成、耳周瘘管或窦道,部分患者同时伴有半侧颜面短小,作为综合征的一部分合并全身其他系统的畸形,需根据不同的畸形类型、患儿发育不同阶段进行专业的序贯性治疗(图1)。
对小耳畸形患者外观和听力学解决方案的制定,目前耳科和整形外科医生的共识为[7]:患者的整体实用听力水平能够进行日常沟通和学习且不伴有外耳道狭窄、胆脂瘤存在及感染风险,首先考虑耳廓重建,避免影响耳廓周围软组织的血供,影响耳廓修复;单侧小耳畸形外耳道闭锁患者一般不影响语言学习,可采用生活中优先照顾等方式进行康复学习;对双侧外耳道完全闭锁患者,出生6个月后可先佩戴软带骨桥,待6岁后行耳再造后或同时行振动声桥、骨桥或其他骨导助听器的植入,考虑到术后耳道狭窄再次闭锁及胆脂瘤形成等情况,不建议行外耳道成形术;对于外耳道狭窄伴发胆脂瘤及感染患者,首先行外耳道成形及中耳手术。小耳畸形外形整复一般采用直埋法(Nagata法)或扩张法行耳廓再造。目前耳廓再造普遍采用肋软骨作为支架,medpor等人工材料,因有远期支架暴露风险,应用比较受限,组织工程软骨支架还在进一步完善中[8,9],对于局部条件无法再造的,也可以使用赝复体恢复外形。大多数医师认为小耳畸形进行耳再造的最小手术年龄为6岁以上,身高>120 cm,剑突水平胸围>55 cm,青春期阶段易出现肋软骨的空泡化,需尽量避开此阶段手术。
另外,小耳患者常伴有面部发育的不对称,轻度面部不对称者,一般无需行颌面部整形。半侧颜面短小畸形(hemifaeial microsomia)患者畸形可累及多个解剖部位,表现为患侧面部短小、腮腺发育不良、咬合面倾斜、颏部偏斜、面横裂、耳畸形及面神经发育不良等[10]。其中骨骼畸形以下颌骨发育不良最常见也最重要,患侧下颌体及下颌升支短小,颞下颌关节发育不良或缺如。严重者可累及上颌骨、颧骨、颧弓及颅颞部骨骼。对于这些患者,一般要根据其年龄、颌畸形程度等进行口腔正畸疗法或骨延长器应用等外科手术治疗,恢复正常的功能和外形。应用下颌骨延长器治疗可与皮肤扩张法再造耳廓同时进行以节省治疗时间。
1.3 单纯性中耳畸形
单纯性中耳畸形指有正常的耳廓,外耳道及鼓膜存在,伴有听骨链或合并面神经发育畸形。按照Teunissen(1993)分类法[11],单纯性中耳畸形可归纳为4类:即先天性镫骨固定;先天性镫骨固定伴听骨链畸形;先天性听骨链畸形但镫骨底板活动;先天性圆窗或前庭窗发育不全或重度发育异常。临床上多表现为自幼或青少年时期发现的单侧或双侧传导性听力障碍,骨导基本正常。不影响生活的单侧传导聋可不予特殊处理,双侧传导聋可以采用手术探查行听力重建或采用气导或骨导助听器提高听力。先天性中耳畸形尤其是伴有前庭窗闭锁患者,约69%同时伴有面神经走形和位置变异,需要在术前充分评估,术中予以高度重视[12]。对于不宜行听骨链重建或重建效果差的病例,如畸形极为严重、面神经走行异常或多次手术不能提高听力者,可配戴常规助听器、骨导助听器或中耳植入装置提高听力。
1.4 内耳畸形
内耳与外中耳来源于不同的胚层,内耳畸形一般单独发生。内耳畸形可发生在骨迷路和膜迷路的任何部分。膜迷路畸形发生在细胞水平,迷路形态无异常改变,影像学方法不能显示,骨迷路畸形因其有特殊的形态学表现可被CT及MRI诊断。关于骨迷路畸形分类,目前土耳其学者Sennaroğlu[13]2017年的内耳畸形的分类方式应用最为广泛,此种分类方法按照耳蜗的发育情况分类:(1)①迷路缺如(Michel畸形);②初级听泡;③耳蜗未发育④共同腔畸形;⑤耳蜗发育不良;⑥不完全分隔 I型、Ⅱ型、Ⅲ型;⑦前庭水管扩大;⑧耳蜗孔异常。(2)蜗神经异常。该分类方法易于影像学分类,为耳鼻喉及相关学人员间对疾病的沟通和理解提供了很好的参考标准。内耳畸形的治疗方案一般根据听力情况和内耳畸形程度进行选择。轻中度听力损失可以配戴助听器,重度极重度听力损失可以根据具体的畸形特点选择相应的电极行人工耳蜗植入,一些特殊类型畸形如Michel畸形、初级听泡、耳蜗未发育、部分耳蜗孔异常、蜗神经异常患者则不适用于人工耳蜗植入,需行听觉脑干植入。
1.5 综合征性耳畸形
国内外已经报道的小耳畸形相关综合征大约有20余种,其中常见的有Treacher Collins综合征,典型症状:颧骨和下颌发育不良、小耳畸形、外耳道闭锁、睑裂外下垂、唇裂等;Branchio-oto-Renal综合征,典型症状:鳃裂囊肿、外耳畸形、听力减退、肾和尿路畸形;Goldenhar综合征,典型症状:眼、耳及脊柱发育异常为主;Nager综合征,典型症状:Treacher Collins综合征伴肢端异常及Miller综合征,典型症状:杯状耳、外侧肢体和面骨发育异常等。外耳位于体表易于观察,对于耳畸形相关综合征的了解,有助于对综合征性疾病的认识和诊治方案的制定。治疗上先行处理危及生命的先天性缺陷,如心脏、血管、气道、尿道等方面的畸形,出生3个月~10岁逐步进行唇腭裂及牙槽突裂的修复解决患儿的喂养问题,耳科需先解决言语学习问题之后进行外观的整复。
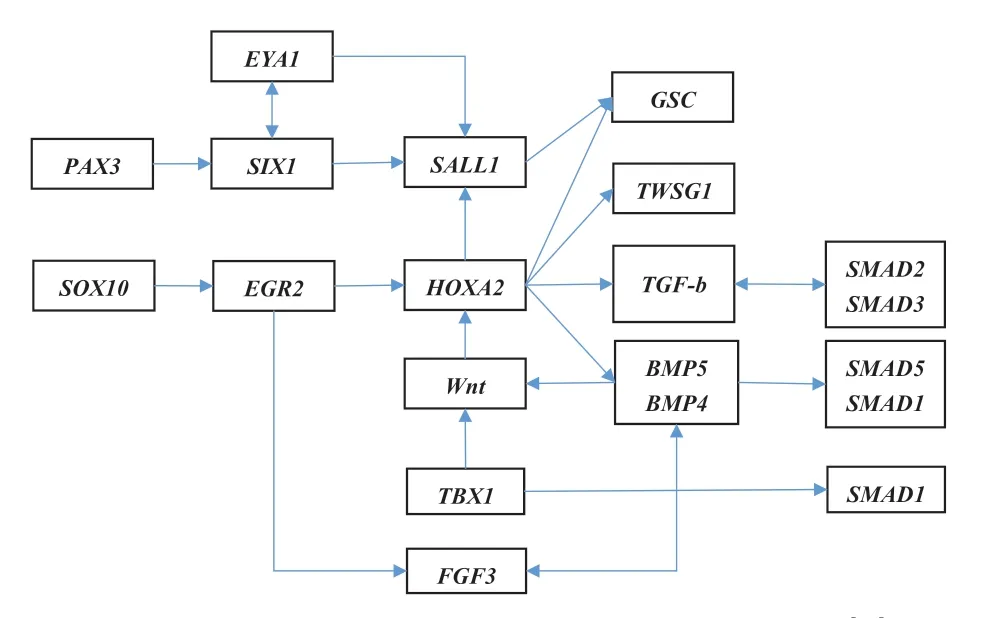
图2 与小耳畸形发生相关的基因调节网络[14]
临床工作中对一些合并中、内耳畸形并具有典型特征的综合征也要有充分认识,如伴甲状腺肿的Pendred综合征,伴有内眦外移、虹膜异色、皮肤及头发色素异常,耳聋或合并有内耳畸形的Waardenburg综合征及伴蓝色巩膜、易骨折、传导性耳聋的Van der Hoeve综合征等。
2 耳畸形的病因学研究及预防
为了有效预防疾病发生,多年来,针对先天性耳畸形病因的研究从未停止,研究者试图从各个层面探讨耳畸形的成因。近年来,神经嵴细胞(neural crest cells,NCC)异常迁徙及以HOXA2为中心影响神经嵴及信号通路的网络作用逐渐引起了人们重视(图2)。章庆国等[15]利用全基因组相关分析(genome wide association study,GWAS)方法对939名中国半侧颜面短小先天性小耳畸形患者和2012名对照人群的90万个遗传变异进行分析,发现ROBO1,GATA3,GBX2,FGF3,NRP2,EDNRB,SHROOM3,SEMA7,PLCD3,KLF12和EPAS1共11个基因存在与13个与小耳畸形显著相关的位点。染色体变异、MicroRNA表达调控异常也与外中耳畸形的发生相关。约2.9%~33.8%的小耳畸形患者有遗传背景[16]。目前虽然部分可能的基因区域和潜在的相关基因被揭示,但耳畸形病因学研究仍是该领域研究的难点。研究显示,小耳畸形的发生可能受环境和基因的共同影响。一方面可能与怀孕早期的异常事件如病毒感染、药物、精神刺激、或父母年龄、辐射、环境污染、种族起源、母亲教育程度、居住环境等有关。但还没有有力的证据说明哪种因素能够起主导作用。目前的数据显示,综合征性及家族性小耳畸形可能与孟德尔遗传有关,散发病例更可能与多因素或多基因相关。
关于内耳畸形,目前确定大前庭水管综合征(90%伴Mondini畸形,即不全分隔Ⅱ型内耳畸形)及不全分隔Ⅲ型内耳畸形分别与SLC26A4和POU3F4基因突变相关,余类型内耳畸形尚未发现明确遗传学相关证据。Waardenburg综合征相关的基因有6个,包括转录因子PAX3、MITF、SNAI2、SOX10和信号分子ENDR3和EDN3。Van der Hoeve综合征属于成骨不全I型,90%以上的患者具有COL1A1基因和COL1A2基因的突变,其中以COL1A1基因改变为主。
综合征性耳畸形里,目前研究比较明确的是Treacher Col1ins综合征,该病多为常染色体显性遗传,外显不全,表现度变异大。63%~93%患者与TCOF1基因变异相关,2%与POL1RC,POL1RD等基因变异相关。2009年,Nature Genetics杂志在线发表了DHODH基因为Miller综合症致病基因的文章,这是首次利用外显子测序技术成功发现一种未知病因的单基因遗传疾病的致病基因报道,开启了利用高通量测序在疾病诊断中的新篇章[17]。近年来,高通量测序技术蓬勃发展,在足够样本量及完整详细临床表型的前提下,全基因组关联分析、外显子组测序、家系连锁分析、拷贝数变异及表观遗传学研究均是小耳畸形病因研究的有效策略。
对于找到遗传学病因的患者和家庭,可以通过遗传咨询、产前诊断或胚胎植入前诊断来预防该家庭生育或再生育严重出生缺陷患者。对于不能明确病因的疾病类型,还需要医务工作者的继续努力。
3 结语
耳畸形表现复杂,不仅严重影响外观和功能,还给患者本人和家庭带来巨大的心理和经济压力,越来越受到社会各界的关注,目前从国家层面也出台了很多与出生缺陷相关的救助政策。因而,深入耳畸形致病机制研究,明确疾病诊疗策略,通过各种途径开展培训工作,提高各级医师的诊疗水平,开展系统化和规范化治疗,有效减少并发症危害、预防疾病的发生,对于实现健康中国的国家战略具有重要意义。
猜你喜欢
中国CT和MRI杂志(2022年7期)2022-06-26
知识就是力量(2021年7期)2021-07-28
中华耳科学杂志(2021年5期)2021-01-02
中华养生保健(2020年3期)2020-11-16
——护肾
饮食科学(2019年2期)2019-11-22
中西医结合心血管病电子杂志(2018年25期)2018-12-14
科技创新导报(2014年20期)2014-11-10
今日中学生(初一版)(2006年8期)2006-07-31
中国美容医学(2004年3期)2004-09-17
为了孩子(孕0~3岁)(2001年12期)2001-07-23
