
HPLC法同时检测人血浆中霉酚酸及其两种代谢产物的浓度
2021-01-05 09:23周金金范国荣
药学服务与研究 2020年6期
周金金,范国荣,3*
[1.广西中医药大学药学院,南宁 530200;2.上海交通大学附属第一人民医院临床药学科,上海200080;3.海军军医大学药学院药物分析学教研室,上海市药物(中药)代谢产物研究重点实验室,上海 200433]
霉酚酸(mycophenolic acid,MPA)是临床常用免疫抑制剂吗替麦考酚酯的活性代谢产物,因其肝毒性、肾毒性低,耐受性好,已被广泛应用于肾、肝或其他组织器官移植后的排异反应和某些自身免疫性疾病的治疗[1]。有研究表明, MPA的药动学和药效学存在极大的个体差异,浓度与疗效之间个体差异很大,浓度与药品不良反应(ADRs)之间也有相关性[2]。而通过治疗药物监测(therapeutic drug monitoring,TDM)调整用药剂量,实现个体化精准用药,可以进一步优化MPA的使用,较好地改善患者预后。
MPA的代谢产物主要为霉酚酸葡萄糖苷(7-O-mycophenolic acid-glucuronide,MPAG)和酰基霉酚酸葡萄糖苷(acyl-mycophenolic acid-glucuronide,AcMPAG)。有研究表明,MPAG本身无药理活性,但在短暂的肝肠循环作用下重新形成MPA,使MPA暴露量增加,导致毒性反应的发生[3-4];另一代谢产物AcMPAG在体内堆积过多时也会引发血液疾病、胃肠道功能紊乱、感染等ADRs[5]。代谢产物的血药浓度与ADRs的关系备受关注,因此,同时检测MPA的代谢产物MPAG、AcMPAG的浓度,评价相关ADRs很重要。目前已有的检测方法有酶放大免疫(enzyme-multiplied immunoassay technique,EMIT)法[6]、HPLC法[7]、LC-MS/MS法[8]和毛细管电泳法[9]等,但关注点往往集中在霉酚酸或者一种代谢产物上。本研究以艾司唑仑为内标,建立了能同时测定人血浆中MPA、MPAG、AcMPAG的HPLC法,并应用于患者血药浓度监测,为临床合理使用吗替麦考酚酯等药物提供参考。
1 材 料
1.1 仪器 Ultimate 3000高效液相色谱仪(美国Thermo公司,包括U3000-VWD检测器和Chromeleon工作站);HY-5涡旋混合器(美国SI公司);Centrifuge 5804 R冷冻离心机(德国Eppendorf公司);FE20型实验室pH计(瑞士Mettler Toledo公司);CPA225D十万分之一电子分析天平(德国赛多利斯公司);SK7200H超声仪(上海科导超声仪器有限公司);Hi-Tech水纯化系统(上海和泰仪器有限公司)。
1.2 试剂 MPA对照品(纯度98%,批号24280-93-1,上海安谱实验科技有限公司);MPAG(纯度98.36%,批号31528-44-6)、AcMPAG对照品(纯度95%,批号99043-04-6,加拿大Toronto Research Chemicals公司);艾司唑仑对照品(纯度98%,批号171219-0102,大连美仑生物技术有限公司);甲醇、乙腈(色谱纯,美国Merck公司);偏磷酸(HPO3,分析纯,国药集团化学试剂有限公司);磷酸二氢钠(分析纯,上海凌峰化学试剂有限公司)。空白血浆采自上海交通大学附属第一人民医院的健康志愿者。
2 方法和结果
2.1 色谱条件 色谱柱为Welch Xtimate C8柱(250 mm×4.6 mm,5 μm)。流动相为甲醇(A)-10 mmol/l NaH2PO4(pH 3.0)水溶液(B),梯度洗脱:0~15 min,46%~30% B,15~20 min,30%~40% B。流速为1.0 ml/min,检测波长215 nm,柱温40 ℃,进样量20 μl。
2.2 对照品溶液与工作液的配制 精密称取MPA、MPAG、AcMPAG、艾司唑仑对照品,用甲醇溶解、稀释并定容,得到浓度分别为1.00、2.50、1.00、1.00 mg/ml的储备液,于-20 ℃保存备用。将上述储备液用50%甲醇逐级稀释得到工作液。工作液中MPA、AcMPAG浓度均为1.96、3.91、7.82、15.63、31.25、62.50、125.00、250.00 μg/ml,MPAG浓度为3.91、7.82、15.63、31.25、62.50、125.00、250.00、500.00 μg/ml,于-20 ℃保存备用。另配制MPA、AcMPAG浓度均为2.50、25.00、200.00 μg/ml及MPAG浓度为5.00、50.00、400.00 μg/ml的低、中、高浓度混合质控工作液,于-20 ℃保存备用。艾司唑仑内标溶液的浓度为20.00 μg/ml,于-20 ℃保存备用。
2.3 血浆样品前处理 精密吸取10 μl工作液,置于1.5 ml Eppendorf管中,加入90 μl空白血浆,再加入10 μl浓度为20 μg/ml的内标溶液,涡旋振荡30 s,加入50 μl 10% 偏磷酸后混匀,再加入50 μl乙腈进行蛋白沉淀,涡旋振荡1 min,以2.081 7×g离心5 min(4 ℃),取上清进样分析,进样体积20 μl。
2.4 专属性实验 取6份不同来源的空白血浆,按2.3项下方法进行样品前处理,按2.1项下色谱条件进行检测,观察色谱图的出峰情况,考察内源性物质及其他物质是否对待测物有干扰。结果在本研究条件下,MPAG、AcMPAG、内标艾司唑仑及MPA的出峰时间分别为4.623、6.627、9.007和10.977 min,血浆中的内源性物质对测定无干扰,联用药物如他克莫司、泼尼松对MPA、MPAG、AcMPAG及内标的测定无干扰,见图1。
2.5 标准曲线的绘制 取10 μl 2.2项下混合工作液,加入90 μl空白血浆中,配制成MPA、AcMPAG浓度为0.196、0.391、0.782、1.563、3.125、6.25、12.5、25.0 μg/ml和MPAG浓度为0.391、0.782、1.563、3.125、6.25、12.5、25.0、50.0 μg/ml的标准曲线样品,按2.3项下方法操作后进样分析,记录峰面积。以待测样品峰面积与内标峰面积的比值为纵坐标(Y),浓度为横坐标(X)进行线性回归。结果MPA和AcMPAG的线性范围为0.19~25.0 μg/ml;MPAG的线性范围为0.39~50.0 μg/ml。MPA的标准曲线方程为Y1=0.519 4X1+0.049,r=0.999 1;MPAG的标准曲线方程为Y2=0.455 5X2+0.503,r=0.999 4;AcMPAG的标准曲线方程为Y3=0.272 2X3+0.006 4,r=0.999 8,表明线性关系良好。
2.6 检测限与定量下限实验 将工作液用50%甲醇多次稀释后,按2.3项下方法进行样品前处理,再进样检测,得到S/N>3的浓度,将S/N>3的浓度定义为检测限浓度(LOD);将标准曲线工作液最低浓度定义为定量下限浓度(LLOQ)。 结果MPA、AcMPAG的检测限为0.048 μg/ml,MPAG的检测限为0.097 μg/ml;MPA、AcMPAG的定量下限为0.19 μg/ml,MPAG的定量下限为0.39 μg/ml。
2.7 精密度和准确度实验 用空白血浆配制MPA、AcMPAG浓度均为0.25、2.5、20.0 μg/ml和MPAG浓度为0.5、5.0、40.0 μg/ml的质控样品,每个浓度5份样品,按2.3项下方法处理后进样测定。每天测定一次,连续测定3 d,记录峰面积,计算批间和批内精密度与准确度,分别用RSD、RE表示。结果日内、日间精密度RSD均<15%,准确度RE均在85%~110%,见表1。
2.8 提取回收率实验 取2.7项下配制的低、中、髙浓度的质控样品,按2.3项下方法处理后进行测定。配制相同浓度的不含生物基质的对照品溶液进行测定,比较峰面积,计算提取回收率。结果MPA、MPAG、AcMPAG和内标艾司唑仑的提取回收率分别为92.50%~105.12%、90.58%~104.82%、86.37%~103.57%、98.11%~101.64%,RSD均<15%(n=5)。

图1 人血浆中霉酚酸及其代谢产物的HPLC谱图A:空白血浆;B:空白血浆+霉酚酸(MPA)+霉酚酸葡萄糖苷(MPAG)+酰基霉酚酸葡萄糖苷(AcMPAG)+ 内标艾司唑仑(浓度分别为25.0、50.0、25.0、20.0 μg/ml);C:患者血浆样品;D:空白血浆+他克莫司(25.0 μg/ml); E:空白血浆+泼尼松(25.0 μg/ml);1:MPAG;2:AcMPAG;3:内标艾司唑仑;4:MPA

表1 血浆中MPA、MPAG、AcMPAG的 精密度和准确度实验结果 (n=5)
2.9 稳定性实验 配制同2.7项下低、中、髙浓度的质控样品,于室温放置6 h、进样盘放置6 h、反复冻融3次、密封低温(-40 ℃)保存1个月,按2.3项下方法处理后进样测定,考察不同条件下样品的稳定性。结果样品在上述条件下均保持稳定。
2.10 临床患者血药浓度监测 国内外均推荐MPA治疗谷浓度为1.0~3.5 μg/ml[10]。但因存在个体差异、药物相互作用等影响因素,MPA谷浓度暴露量与推荐治疗窗有差异,因此应对MPA进行治疗药物监测。
本研究收集2017-2018年上海交通大学附属第一人民医院肾移植术后复查的门诊患者28例,均长期服用麦考酚钠肠溶片或吗替麦考酚酯胶囊和其他免疫抑制剂。抽取患者谷浓度血样,检测MPA血药浓度和生化指标。结果MPA血药浓度在0.154~6.65 μg/ml范围,见表2。其中有16例患者MPA谷浓度不在1.0~3.5 μg/ml范围内,与推荐治疗范围有差异,但患者肝、肾功能指标均在正常范围,身体机能处于稳定状态,未发生ADRs,因此维持用药剂量不变。考虑到患者由于个体间代谢差异、饮食等因素影响,是否调整用药剂量和用药方案确保机体各指标正常,需继续进行临床TDM。
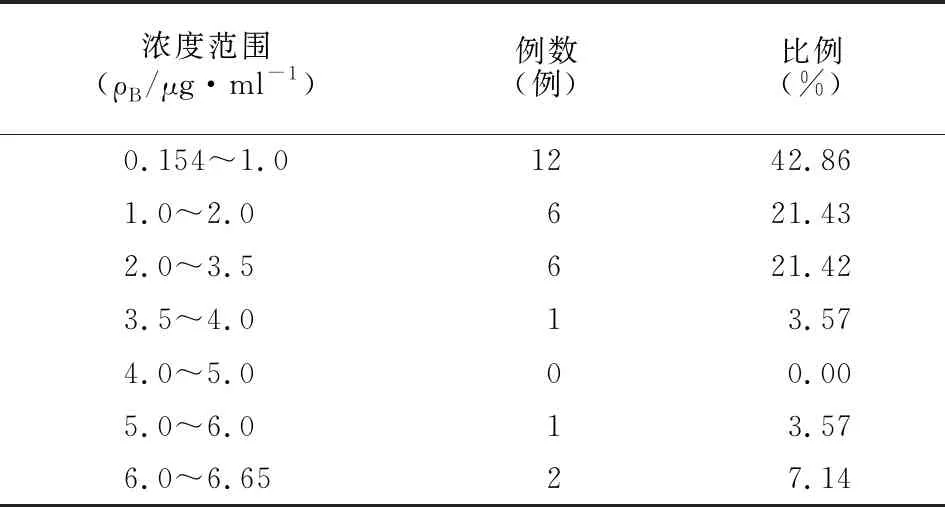
表2 28例肾移植患者霉酚酸血药浓度分布范围
3 讨 论
3.1 色谱条件的考察 在选择流动相时,本研究考察了5、10、15 mmol/L NaH2PO4对各待测物出峰的影响,并且在酸碱度的选择上,为契合各化合物的性质,使待测物在洗脱过程中稳定存在,不黏附,经优化后最终选择了10 mmol/L NaH2PO4(pH 3.0)作为水相。
本研究参考国内外文献,研究了不同型号色谱柱如Waters Symmetry C18柱(150 mm×4.6 mm,5 μm)、Waters XBridge C18柱(150 mm×4.6 mm,3.5 μm)、Welch Xtimate C8柱(250 mm×4.6 mm,5 μm)、Agilent ZORBAX SB-AQ柱(150 mm×4.6 mm,5 μm)、Waters Atlantis T3柱(100 mm×3 mm,3 μm),发现待测物在填充粒径为5 μm、内径为4.6 mm的色谱柱中出峰结果较好。待测物在250 mm柱长的色谱柱中能更快出峰,从而节省了总的运行时间。代谢产物MPAG、AcMPAG极性较高,含有的基团增加了其亲水性,不同填充材料和不同键合相对色谱分离也有影响。AQ、C18色谱柱和T3色谱柱在洗脱化合物时也能达到分离的目的,但MPA和MPAG、AcMPAG极性相差较大,且调节流动相发现,MPA对水相的变化较敏感,不能同时满足时间短、分离度好的要求,而使用Welch Xtimate C8色谱柱,在40 ℃的柱温、合适的流动相条件下具有较好的分离度,峰型也比较好,在12 min内4种物质均能出峰,满足检测的要求。
本研究在200~600 nm波长扫描后发现,MPA、MPAG、AcMPAG在215、254、305 nm波长下均有紫外吸收,代谢产物MPAG、AcMPAG在215 nm波长下吸收更强。与文献[11]中报道的使用UV检测MPA、AcMAG,荧光检测MPAG,使用双内标的方法相比较,本研究采用215 nm检测波长,灵敏度高,内源性物质无干扰,能同时检测3种待测物及内标。
3.2 样品前处理条件的考察 文献报道的血浆样品前处理方法有固相萃取法[12]、液-液萃取法[13]和蛋白沉淀法[6]。本研究比较这3种样品前处理方法。固相萃取实验中使用的萃取小柱消耗高、成本高;在液-液萃取实验中,本研究考察了不同的有机溶剂乙酸乙酯、叔丁基甲基醚、异丙醇、二甲亚砜对待测物回收率的影响,发现液-液萃取法中待测物的回收率不足40%,且过程耗时长;在蛋白沉淀法实验中,通过对比用硫酸锌、钨酸钠、高氯酸、偏磷酸、乙腈、甲醇对样品进行前处理后发现,同时加入乙腈和10%偏磷酸前处理样品,待测物的回收率>70%,准确性好,且操作简便、快速,能满足检测要求。本研究经过考察,最终选用同时加入乙腈和10%偏磷酸前处理样品的蛋白沉淀法。
3.3 内标的选择 本研究考察了不同的内标如阿立哌唑、苯妥英钠、艾司唑仑、氯丙嗪等,发现艾司唑仑不干扰待测物,响应好,因此选用艾司唑仑作为内标。
本研究建立了一种简便、可靠、能同时检测人血浆中MPA、MPAG、AcMPAG浓度的HPLC法,可用于患者MPA血药浓度监测,为临床合理、安全、有效用药提供技术支持,进一步保障用药安全。
猜你喜欢
中国氯碱(2022年10期)2022-11-22
健康体检与管理(2022年4期)2022-05-13
化学工程师(2020年7期)2020-09-07
中国医药指南(2020年17期)2020-07-21
中国外汇(2019年10期)2019-08-27
中国外汇(2019年10期)2019-08-27
中国外汇(2019年22期)2019-05-21
中国外汇(2019年21期)2019-05-21
制造技术与机床(2017年9期)2017-11-27
中华胃食管反流病电子杂志(2017年2期)2017-10-27
