
大肠杆菌双质粒CRISPR-Cas9系统的优化及应用
2021-02-10 07:37王凯凯王晓璐苏小运张杰
生物技术通报 2021年12期
王凯凯 王晓璐 苏小运 张杰
(中国农业科学院北京畜牧兽医研究所 动物营养学国家重点实验室,北京 100193)
CRISPR-Cas9是十分便捷、有效的基因组编辑工具[1],近年来已经成功应用于植物、动物、微生物等多种生物中。Xing等[2]在pGreen和pCAMBIA载体上设计组装多个gRNA序列,引导Cas9蛋白同时特异性识别和切割多个基因序列,在玉米和拟南芥等植物中实现了基因高效编辑。Li等[3]以Tet家族基因作为靶标,经显微注射将靶向Tet的sgRNA与Cas9 mRNA一起引入大鼠胚胎细胞中完成了多基因高效编辑。甲基营养酵母巴斯德毕赤酵母是生产异源蛋白质最常用的表达系统之一,经密码子优化的Cas9基因序列与不同启动子组成表达盒,可实现接近100%编辑效率[4]。在大肠杆菌中,Zhao等[5]通过Golden Gate方法将Cas9系统与促进同源重组的蛋白基因recA组装,成功构建了pRed_cas9_recA_Δpoxb300质粒,利用此单质粒对lacZ基因敲除验证了敲除效率,并且利用其携带的温敏性复制起始位点通过提高培养温度剔除质粒可进行连续基因敲除。
理论上CRISPR-Cas9系统可以靶向任何基因进行编辑,但实验发现微生物中关键基因的敲除依然十分困难。CRISPR-Cas9系统针对不同的基因或宿主时,基因编辑往往会受到十分复杂的影响,除了gRNA选择的影响外,基因组编辑效率还受到非同源末端连接(non-homologous end joining,NHEJ)和同源重组(homology directed repair,HDR)DNA修复机制的影响[6]。前期关于CRISPR-Cas9系统已有大量报道提供了优化策略,以提高基因编辑效率和避免脱靶效应[7-8],但依然未能解决关键基因敲除效率下降的问题。在微生物的代谢改造中,根据设计需求往往需要对影响宿主生长及代谢的关键靶点基因进行单个或连续的基因编辑,以改变宿主的生长状态或代谢流流向,达成代谢改造目的[9-10]。但由于某些基因的特殊性以及突变体的代谢特性变化等,在编辑关键基因的过程中会出现CRISPR-Cas9系统的“失灵”,造成编辑效率极低或者不能完成编辑[11]。gltA为柠檬酸合酶编码基因,是控制代谢流进入三羧酸循环的关键酶,敲除可严重降低大肠杆菌的生长速率[12],Heo等[13]对gltA基因的编辑效率仅为30%左右,低于其他基因(frdA等)的编辑效率。kefB基因编码受谷胱甘肽调节的钾外排系统蛋白KefB[14],可防止钾离子与亲电试剂的结合,催化K+/H+逆浓度跨膜,对于细胞信号传导以及维持细胞的渗透压具有重要的作用,Jiang等[15]尝试通过CRISPR-Cas9系统对此基因进行改造,经同源臂长度等优化依然未能成功获得基因缺失的突变株。由此可见,对于大部分基因,包括那些本身对细胞生长和代谢平衡影响较小的基因以及功能可被替代的基因进行编辑时,均可通过优化编辑过程获得较高的编辑效率。但对细胞生存具有重大意义的基因进行编辑时往往比较困难,因此在编辑宿主的关键基因时需要根据实际需求优化基因编辑系统,以提升编辑效率。
大肠杆菌中,磷酸果糖激酶(pfk编码)是糖酵解途径的主要限速酶之一,包含由pfkA、pfkB两个基因编码的同工酶I和同工酶II,可依靠ATP提供的磷酸基团催化果糖6-磷酸生成果糖1,6-二磷酸[16]。果糖1,6-二磷酸属于糖酵解途径的主要中间代谢产物之一,决定进入糖酵解ATP和NADH产生阶段的代谢流的多少。另外,葡萄糖的另一代谢去路是经6-磷酸葡萄糖脱氢酶(zwf编码)催化进入磷酸戊糖途径[17]。6-磷酸葡萄糖脱氢酶是葡萄糖进入磷酸戊糖途径的第一个催化酶,催化6-磷酸葡萄糖氧化为6-磷酸葡萄糖酸内酯,控制着整个磷酸戊糖途径的代谢通量。有研究表明,通过zwf基因的缺失可促进甘油的利用[18]。甘油代谢是由甘油激酶(glpK编码)在ATP存在下催化甘油生成3-磷酸甘油后进入糖酵解途径,该催化反应是甘油代谢的限速步骤,对于甘油的利用至关重要[19]。此前研究表明,pfk、zwf基因缺失及glpK基因过表达的突变体在碳源利用试验中表现出十分明显的代谢表型差异。前期工作中,我们尝试利用Zhao等[5]建立的单质粒CRISPR-Cas9系统进行pfk、zwf基因的敲除及glpK基因的敲入,但编辑效率很低,且有的基因无法获得突变株。因此在本研究中优化了双质粒CRISPR-Cas9系统,成功获得了pfkB敲除及glpK敲入的突变株,且提高了pfkA、zwf基因的编辑效率。对获得的突变株利用碳源的效率进行了验证,进一步确认了基因缺失与突变体代谢能力的关系。本研究优化得到的双质粒CRISPR-Cas9系统为关键基因编辑效率较低的问题提供了可行性解决方案。
1 材料与方法
1.1 材料
大肠杆菌Top10为克隆宿主,用于质粒构建;大肠杆菌MG1655(DE3)为代谢改造出发菌株;pEasy-T3质粒购自北京全式金公司,用于构建携带gRNA和同源臂的编辑质粒;pRed_cas9_recA_Δpoxb300质粒购自MolecularCloud,是Cas9表达载体。Sugar-ParkI,10 μm,6.5 mm×300 mm色谱柱购自Waters公司,用于分析发酵底物葡萄糖和甘油的消耗。DNA聚合酶、重组酶、质粒小提中量试剂盒等相关的酶均购自Vazyme公司;卡那霉素、氨苄青霉素钠购自北京索莱宝科技有限公司;其余试剂购自上海国药集团化学试剂有限公司。A20高效液相色谱仪购自日本岛津公司;凝胶成像仪购自美国Bio-Rad公司;其余菌体培养摇床及恒温培养箱均为国产设备。
1.2 方法
1.2.1 菌株培养 大肠杆菌克隆宿主Top10和大肠杆菌MG1655(DE3)在LB中培养,培养条件为37℃,200 r/min,抗生素根据质粒携带标记基因进行添加,卡那霉素添加浓度为50 μg/mL,氨苄青霉素为 100 μg/mL ;携带有 pRed_cas9_recA_Δpoxb300质粒的宿主MG1655-pox300在30℃培养,培养基中添加卡那霉素;在MG1655-pox300宿主基础上导入靶基因敲除质粒的宿主MG1655-pox300-pΔpfkA、MG1655-pox300-pΔpfkB、MG1655-pox300-pΔzwf、MG1655-pox300-pΔABE∷glpK在 30℃ 培 养, 培 养基中添加卡那霉素和氨苄青霉素。经基因编辑的菌株 MG1655-ΔpfkA、MG1655-ΔpfkB 和 MG1655-Δzwf在M9培养基中发酵,MG1655-ΔABE∷glpK菌株在MK5(含0.5%甘油且无葡萄糖的M9培养基)和MK105(含1%葡萄糖和0.5%甘油的M9培养基)中培养,验证突变株对碳源的利用情况。
1.2.2 质粒构建与转化 从MG1655(DE3)大肠杆菌基因 zwf、pfkA、pfkB、nagABE中选取靶向20 nt序 列( 分 别 为 pfkA 5′-gtgtctgacatgatcaaccg-3′;pfkB 5′-cacgtacatgtggaagcaag-3′;zwf 5′-gcgtgctgactgggataaag-3′;nagABE 5′-cctgcagcgccagtgctttc-3′), 设计引物并以pRed_cas9_recA_Δpoxb300质粒为模板扩增出gRNA scaffold基因片段,启动子J23119与gRNA scaffold序列通过overlap PCR连接在一起启动gRNA的表达,获得的连接片段与pEASY-T3载体使用T4连接酶连接,连接产物导入大肠杆菌克隆宿主Top10中,37℃培养,PCR筛选阳性菌落,获得质 粒 pΔpfkA-gRNA、pΔpfkB-gRNA、pΔzwf-gRNA 和pΔABE-gRNA。使用Sbf I、Nde I限制性内切酶37℃处理上述质粒2 h,凝胶电泳回收载体片段,并与从MG1655(DE3)基因组中扩增的基因上下游同源臂片段(~500 bp)混合,使用2×Express clone试剂盒(诺唯赞)在50℃连接15 min,转化至大肠杆菌Top10中,在氨苄青霉素抗性平板上筛选阳性转化子 获 得 质 粒 pΔpfkA、pΔpfkB、pΔzwf、pΔABE( 表1)。以毕赤酵母GS115基因组为模板,扩增glpK基因片段,并与通过Xho I、Sbf I限制性内切酶处理的pΔABE质粒片段连接,导入大肠杆菌Top10感受态细胞中,筛选获得质粒pΔABE∷glpK。glpK基因由T7启动子启动表达。大肠杆菌Top10化学感受态细胞外源质粒转化方法参照商品说明(天根,北京),大肠杆菌MG1655(DE3)质粒转化均采用电转化方式,感受态制备参照Yoshida等[20]的方法。本研究所用引物如表2所示。单质粒CRISPR-Cas9系统分别通过PCR方式将gRNA与同源臂序列组装至pRed_cas9_recA_Δpoxb300质粒上,单质粒系统的20 nt靶向序列与双质粒系统相同。构建成功 pRAΔpfkA、pRAΔpfkB、pRAΔzwf和pRAΔABE∷glpK,构建参照Zhao等[5]的方法。

表1 本研究中使用的菌株和质粒Table1 Strains and plasmids used in this study

表2 本研究中使用的引物Table 2 Primers used in this study
1.2.3 基因敲除 双质粒基因敲除的方法如下:首先将pRed_cas9_recA_Δpoxb300质粒通过电转化导入 MG1655(DE3)中获得 MG1655-pox300菌株;其 次 将 构 建 好 的 质 粒 pΔpfkA、pΔpfkB、pΔzwf、pΔABE∷glpK分别转化至MG1655-pox300中,在含有卡那霉素和氨苄青霉素的双抗性LB平板上筛选获得包含有两个质粒的菌株分别命名为MG1655-pox300-pΔpfkA、MG1655-pox300-pΔpfkB、MG1655-pox300-pΔzwf、MG1655-pox300-pΔABE∷glpK,并将上述菌株接种到同样的双抗性LB液体培养基中,30℃培养16 h后涂布于含有2 g/L阿拉伯糖的双抗固体LB平板上,诱导CRISPR-Cas9表达进行基因编辑。筛选平板在30℃条件下培养18 h,通过使用灭菌的牙签随机挑选单菌落,并进行菌落PCR确认基因敲除或敲入的效果。获得的突变菌株在42℃下传代5次丢除所有质粒。
单质粒基因敲除步骤如下:将构建成功的质粒pRAΔpfkA、pRAΔpfkB、pRAΔzwf、pRAΔABE∷glpK分别导入MG1655(DE3)感受态中,在卡那霉素抗性平板中进行基因编辑。具体基因编辑步骤参照Zhao等[5]的方法。
1.2.4 葡萄糖及甘油高效液相色谱(HPLC)检测大肠杆菌MG1655(DE3)、MG1655-ΔpfkA、MG1655-ΔpfkB、MG1655-Δzwf、MG1655-ΔABE∷glpK 使用 M9、MK5、MK105培养基进行发酵,分别在0、3、6、9、12、24、72 h进行取样。葡萄糖、甘油浓度测定采用高效液相色谱法。高效液相色谱仪为岛津20A,使用 Sugar-Park I色谱柱(10 μm,6.5 mm×300 mm),流动相为H2O,流速0.6 mL/min,柱温80℃,上样量10 μL,利用示差检测器RID-20A进行检测。
2 结果
2.1 双质粒CRISPR-Cas9基因编辑系统的构建
大肠杆菌CRISPR-Cas9基因编辑系统主要包括Cas9、gRNA和同源臂3个部分,其中Cas9蛋白起着切割基因组的作用,因此Cas9相对于细胞来说具有一定的毒性,其表达量过大可能会造成细胞的死亡[21]。而gRNA起着引导Cas9蛋白寻找靶位点的作用,同源臂则负责修复被CRISPR-Cas9系统切断的基因组,因此如果两者的浓度过低会造成Cas9无法找到靶位点或切断的基因组无法被修复,由此会造成编辑效率的降低或编辑的失败。通常的研究会把Cas9、gRNA和同源臂3个部分置于一个质粒上(称为单质粒CRISPR-Cas9系统,图1-A),往往会造成三者之间的比例不协调,从而导致基因编辑效率低下。基于此,本研究中拆分Cas9蛋白和同源臂-gRNA单元为两个独立的部分,通过控制Cas9蛋白的表达,并增加同源臂-gRNA浓度的方式进行基因的编辑。Cas9蛋白被置于低拷贝的质粒上(复制子pSC101,拷贝数约为5)[22],并采用诱导表达的方式控制表达量;同源臂-gRNA单元被置于高拷贝的质粒上(复制子pMB1,拷贝数>500)[23],并采用强启动子J23119控制gRNA的表达,以提高gRNA的表达量及同源臂的浓度(图1-B)。携带有Cas9蛋白和携带有同源臂-gRNA单元的两个质粒协同作用便可完成基因组编辑的功能,即为双质粒CRISPR-Cas9编辑系统。
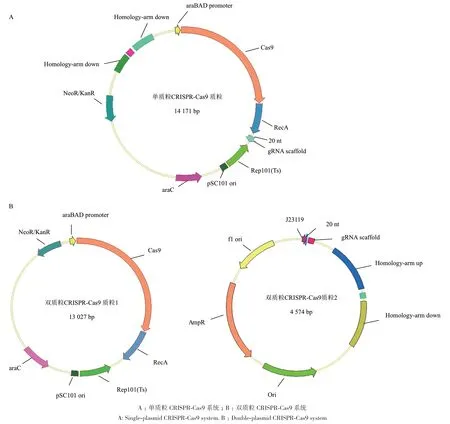
图1 双质粒CRISPR-Cas9系统的优化Fig. 1 Optimization of double-plasmid CRISPR-Cas9 system

续表Continued
2.2 糖酵解途径关键基因的敲除
磷酸果糖激酶pfkA、pfkB是大肠杆菌糖酵解途径中的关键基因,这两个基因的敲除会对大肠杆菌利用葡萄糖的能力产生一定的影响。因此本研究首先选择这两个基因进行敲除验证双质粒CRISPR-Cas9系统的编辑效率。分别选择5′-gtgtctgacatgatcaaccg-3′(pfkA 20 nt)和 5′-cacgtacatgtggaagcaag-3′(pfkB 20 nt)序列作为pfkA和pfkB基因敲除的引导序列,所用同源臂长度均为500 bp左右,以此分别构建了单质粒和双质粒CRISPR-Cas9基因敲除系统。通过检测引物对基因敲除结果进行确认,如表3和图2所示。随机挑选16个克隆子进行敲除效率验证,其中双质粒CRISPR-Cas9系统对pfkA基因敲除中共获得6个阳性克隆子,基因敲除效率为37%;pfkB基因敲除中共获得1个阳性克隆子,基因敲除效率为6%。单质粒CRISPR-Cas9系统可成功敲除pfkA基因,16个随机样本中阳性克隆子为1个,效率仅为6%,而在尝试敲除pfkB基因时未能检测到基因敲除的阳性突变株。由此可见,双质粒CRISPRCas9系统在敲除大肠杆菌糖酵解途径关键基因时效率明显高于单质粒CRISPR-Cas9系统。
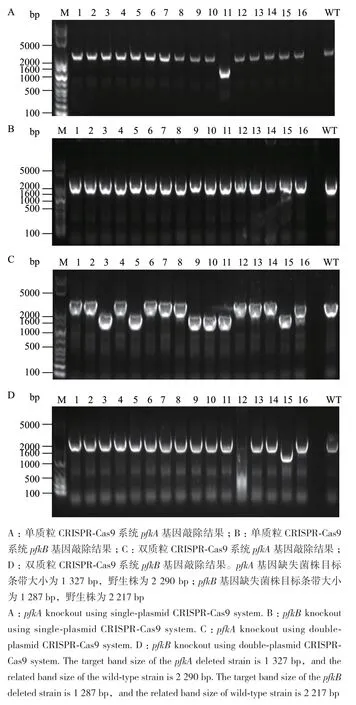
图2 pfkA、pfkB基因敲除电泳结果Fig. 2 Identification of pfkA and pfkB deletion by gel electrophoresis

表3 基因编辑效率比较Table 3 Comparison of gene editing efficiency
2.3 糖酵解基因敲除对细胞葡萄糖代谢的影响
将获得的突变株MG1655-ΔpfkA和MG1655-ΔpfkB在以葡萄糖为唯一碳源的M9培养基中发酵,并表征糖酵解途径关键基因pfkA和pfkB的敲除对菌体代谢葡萄糖的影响(图3-A)。结果表明,敲除pfkA或pfkB基因会对大肠杆菌利用葡萄糖的能力产生不同的影响。pfkA被认为是主要的催化酶,敲除后对突变体葡萄糖分解代谢呈现出较大的影响。与野生株相比,突变株MG1655-ΔpfkA的葡萄糖消耗速率在发酵前期(12 h内)明显降低,但是经过一段时间的适应后,葡萄糖消耗速率迅速提高,最终的葡萄糖总消耗量略高于野生株,72 h消耗量为4 g/L。与葡萄糖的消耗相对应,突变株MG1655-ΔpfkA的生长速率在对数生长期相较于野生株也有所降低(图3-B)。敲除pfkB基因后的突变体MG1655-ΔpfkB的葡萄糖消耗速率相较于野生株也被减缓了,但略高于pfkA缺陷株,最终经72 h培养后菌体消耗葡萄糖量为3.5 g/L,小于野生株的3.9 g/L。
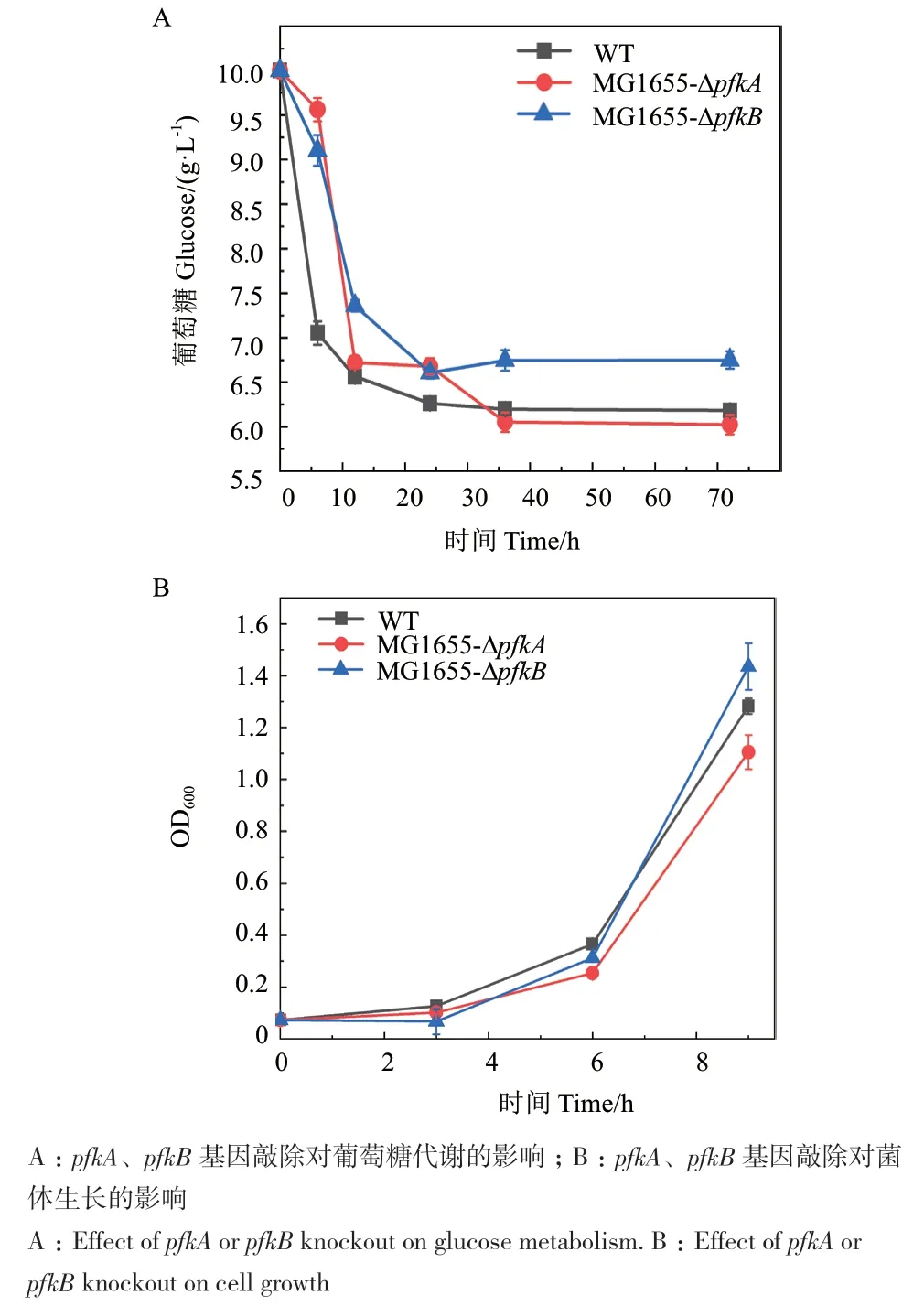
图3 pfkA、pfkB基因敲除菌株表型验证Fig. 3 Verification of the phenotypes of pfkA and pfkB deleted mutants
2.4 磷酸戊糖途径关键基因的敲除
为了进一步验证双质粒CRISPR-Cas9系统的敲除效率,再次选择磷酸戊糖途径中的关键基因zwf进行敲除。5′-gcgtgctgactgggataaag-3′序列被用作zwf敲除的引导序列,同源臂长度同样为500 bp左右,构建单质粒和双质粒CRISPR-Cas9系统。随机挑选8个单克隆进行基因敲除效率的验证,结果显示,双质粒CRISPR-Cas9系统敲除zwf基因的样本全部为阳性克隆子,敲除效率达到100%,而单质粒CRISPR-Cas9系统共获得3个阳性克隆,敲除效率仅为37%(图4和表3)。结果再次证明双质粒CRISPR-Cas9系统在敲除大肠杆菌关键基因时效率优于单质粒CRISPR-Cas9系统。
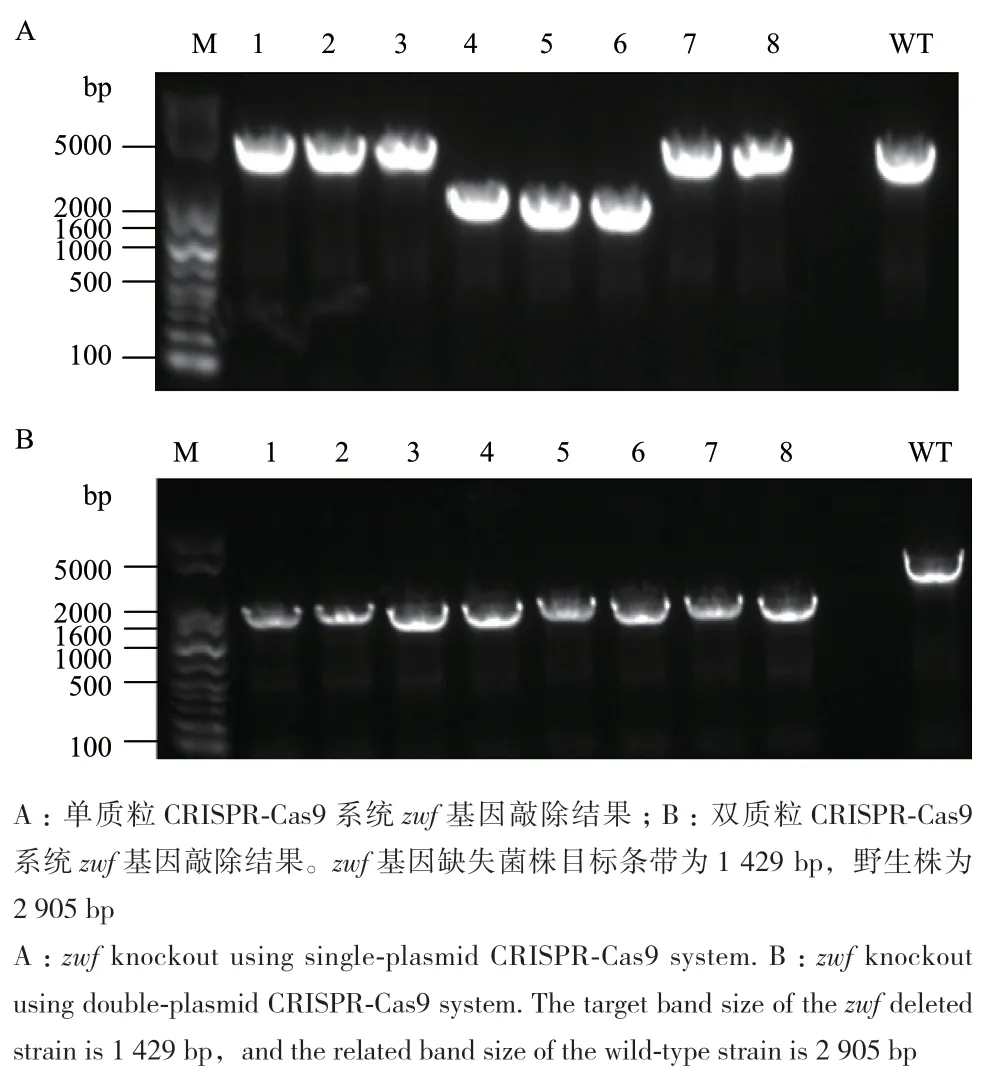
图4 zwf基因敲除电泳结果Fig. 4 Identification of zwf deletion by gel electrophoresis
2.5 磷酸戊糖途径关键基因敲除对葡萄糖代谢的影响
敲除zwf基因的缺陷型菌株MG1655-Δzwf在以葡萄糖为唯一碳源的M9培养基中的生长受到严重的阻碍,葡萄糖消耗速率降低了大约32%,经72 h发酵葡萄糖消耗总量降低了1.2 g/L(图5-A)。缺失zwf基因的突变株生长也受到了较大程度的影响,与葡萄糖消耗保持相似的规律(图5-B)。zwf基因的缺失阻碍了磷酸戊糖途径的下游代谢,导致NADPH合成受阻,其参与的代谢活动被抑制。NADPH在大肠杆菌代谢活动中的全局参与度较高,因此在其合成受到抑制时造成对葡萄糖分解代谢整体的影响较大。

图5 zwf基因敲除菌株表型验证Fig. 5 Characterization of the phenotypes of zwf deleted mutant
2.6 基因敲入效率
除了基因敲除外,对双质粒CRISPR-Cas9系统的基因敲入效率也进行了验证。在大肠杆菌基因组中,nagA、nagB、nagE连续排列,通过选择靶向于nagB 基因的引导序列 5′-cctgcagcgccagtgctttc-3′,选用nagA基因上游和nagE下游的长度约500 bp的序列作为同源臂,并在同源臂中间添加来源于毕赤酵母的glpK基因,构建单质粒和双质粒CRISPR-Cas9基因替换系统。将构建成功的质粒pΔABE∷glpK导入MG1655-pox300中,依照建立的双质粒CRISPRCas9系统突变株筛选流程进行基因替换。如图6所示,使用双质粒CRISPR-Cas9系统在nagABE基因位点整合glpK,在10个随机样本中共获得1个阳性克隆子,基因敲入效率为10%。与之相对比,使用单质粒CRISPR-Cas9系统时glpK基因未能整合至MG1655(DE3)的基因组中。结果说明,双质粒CRISPR-Cas9系统在大肠杆菌基因敲入方面也具有一定的优势。
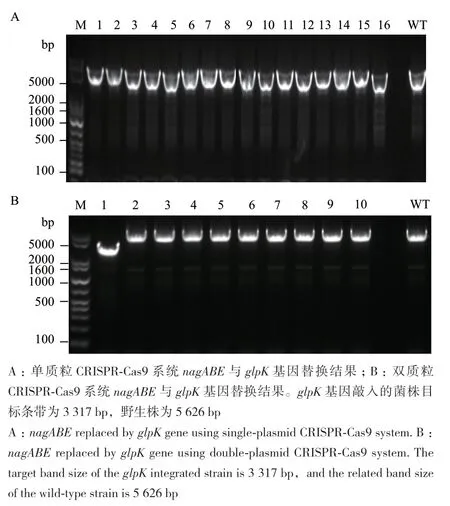
图6 nagABE与glpK基因替换电泳结果Fig. 6 Identification of nagABE replaced by glpK via gel electrophoresis
2.7 MG1655-ΔABE∷glpK突变株的甘油利用效率
通过将来源于毕赤酵母GS115的甘油激酶基因glpK整合至大肠杆菌MG1655(DE3)的nagABE位点,获得MG1655-ΔABE∷glpK突变株。在以甘油为唯一碳源或甘油、葡萄糖混合碳源的两种培养基中测定MG1655-ΔABE∷glpK突变株的甘油代谢状态,并绘制甘油代谢曲线如图7所示。混合碳源MK105(含葡萄糖和甘油混合碳源)中MG1655-ΔABE∷glpK突变株的甘油消耗速率远大于野生株,野生株中的甘油利用受到强烈的葡萄糖抑制。而整合甘油激酶的大肠杆菌MG1655-ΔABE∷glpK受到葡萄糖的抑制作用减弱,在72 h发酵中消耗了0.8 g/L甘油。在MK5培养基中(以甘油为唯一碳源),MG1655-ΔABE∷glpK对甘油的代谢能力相比于野生株提高了30%,72 h消耗甘油量为3.2 g/L(图7)。
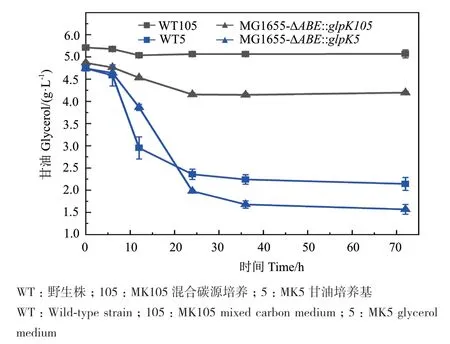
图7 MG1655-ΔABE∷glpK突变株甘油利用状况Fig. 7 Glycerol utilization by MG1655-ΔABE∷glpK mutant
3 讨论
近年来,CRISPR-Cas9基因编辑工具及其突变体如nCas9、dCas9等[24-25]因其在基因编辑及基因表达抑制中特异性较高、编辑效率高及编辑周期短等特点被广泛应用于多种生物中[26-28]。对于CRISPR-Cas9系统的优化已经有许多文献的报道,通过设计合理的gRNA、延长同源臂序列长度以及额外表达λ-red等促进同源重组的蛋白都可以提高基因编辑效率[29]。李秋月等[30]利用在线软件设计gRNA并通过体外检测筛选出最优的gRNA用于载体的构建,在番茄子叶中显著提高了CRISPR-Cas9的编辑效率。Baker等[31]为了优化CRISPR-Cas9系统的编辑效率,在小鼠胚胎干细胞中使用次黄嘌呤-鸟嘌呤磷酸核糖基转移酶测定法测量了Cas9系统的编辑效率与同源臂长度之间的关系。研究表明,CRISPR-Cas9系统中基因的编辑效率随着同源臂长度的增加而提高,与0.4 kb同源臂相比,在4 kb同源臂下可使基因编辑效率提升约5倍,同源臂长度为20 kb时编辑效率提升约10倍。Su等[32]利用CRISPR-Cas9系统在大肠杆菌代谢改造中通过λ-Red辅助的同源重组,将最大12 kb的DNA模块整合到大肠杆菌W3110的染色体中。该方法的效率可以达到100%,显着提高了外源基因敲入的效率。
虽然通过一些优化策略可以提高CRISPR-Cas9系统的编辑效率,但不得不承认,即使是针对大肠杆菌这种广泛应用于生化实验中的模式宿主菌,CRISPR-Cas9系统也难以保证对基因组中的所有基因都可以高效编辑。由于某些基因在维持宿主生存或重要代谢过程中发挥极其重要的作用,因此这些基因的编辑效率相比其他非关键基因会有很大程度的降低,甚至不能编辑。本研究从增加gRNA的表达及同源臂在菌体中浓度的角度出发,将CRISPRCas9系统拆分成Cas9蛋白和同源臂-gRNA单元两个部分,分别组装至低拷贝质粒和高拷贝质粒上,进行非等浓度的表达。这样既可以降低Cas9蛋白过量表达对细胞造成的毒害作用[33],又可以通过饱和的gRNA和同源臂完成靶位点的寻找、切割及重组。试验结果证明,在敲除糖酵解和磷酸戊糖途径中的关键基因时,双质粒CRISPR-Cas9系统的基因敲除效率明显优于单质粒系统。而且在大肠杆菌基因组整合试验中,双质粒CRISPR-Cas9系统也具有一定的优势。这些结果均说明,提高引导Cas9蛋白对DNA切割的gRNA的表达量和维持过量的同源臂片段有利于筛选获得突变株。前期的实验也表明,以卡那霉素维持菌体内的Cas9蛋白表达质粒pRed_cas9_recA_Δpoxb300,可轻易通过连续传代在完成无痕基因编辑后去除同源臂-gRNA供体质粒。因此,在保持pRed_cas9_recA_Δpoxb300质粒的同时,通过更换携带有不同gRNA和同源臂的供体质粒即可完成连续的基因编辑。这样在提升编辑效率的同时也大大降低了代谢改造的工作量和操作难度,为高效构建大肠杆菌工程菌株奠定了基础,也为在其他微生物中建立更加高效、便捷的基因编辑方法提供了借鉴。
4 结论
本研究通过提高CRISPR-Cas9系统中gRNA的表达量及同源臂在菌体中的浓度,在大肠杆菌中优化了双质粒CRISPR-Cas9系统。利用该系统高效敲除了糖酵解途径关键基因pfkA、pfkB和磷酸戊糖途径关键基因zwf,并成功将甘油激酶glpK基因整合到nagABE基因簇位点。与单质粒CRISPR-Cas9系统相比,优化的双质粒CRISPR-Cas9系统无论在关键基因的敲除或外源基因的敲入方面均具有更高的效率。
猜你喜欢
基层中医药(2022年4期)2022-07-22
生物化工(2021年6期)2022-01-27
磷肥与复肥(2021年8期)2021-09-28
汉字汉语研究(2021年2期)2021-08-30
无机盐工业(2020年11期)2020-11-21
中国化肥信息(2019年12期)2020-01-16
汉字汉语研究(2019年2期)2019-08-27
能源(2017年7期)2018-01-19
新高考·英语进阶(高二高三)(2018年8期)2018-01-15
中学科技(2017年11期)2017-12-26
