
磷化钴材料在电化学能源领域的研究进展
2021-04-20 10:30赵鹬周飞张伟伟李宁李世友李贵贤
化工进展 2021年4期
赵鹬,周飞,张伟伟,李宁,李世友,李贵贤
(兰州理工大学石油化工学院,甘肃兰州730050)
过渡金属磷化物兼具金属的电子性质和陶瓷的物理特性,不仅有较高的化学稳定性和热稳定性,同时还是热和电的良导体[1]。一方面,过渡金属磷化物表面能可逆地吸附/解吸氢,促进加氢过程;另一方面,过渡金属磷化物具有缺陷和变形的晶体结构可以很好地插入Li+和Na+等离子,从而可成为优异的电极材料[2]。过渡金属磷化物已被广泛用于众多的绿色能源领域,主要包括:①传统能源精制处理的新型催化剂,如对馏分油中的噻吩类和含氮杂环化合物进行加氢处理,以去除硫、氮等易产生有害物质的元素,合成清洁能源[3-4];②生物油的优化催化剂,如木质纤维素等生物质快速热解能产生大量可再生的生物油,然而生物油中含氧化合物含量较高,导致产物本身性能不理想(黏度大、腐蚀性强、产热低等),利用磷化物催化加氢脱氧是极有前景的优化方法之一[5-6];③更高效的电化学能源的电极材料或催化剂,在锂/钠离子电池、燃料电池、超级电容器、水解制氢等领域发挥优异的作用[7-8]。作为过渡金属磷化物家族重要的一员,磷化钴材料同样备受关注。早在20世纪80年代,Nozaki等[9]便通过程序升温还原制备了负载在Al2O3上的Co2P,并将其用于1,3-丁二烯加氢。其后一段时间磷化钴大多作为加氢催化剂进行研究。在对加氢性能的深入研究中,研究者发现磷化钴对噻吩加氢脱硫(HDS)的活性非常高,并且更重要的是其中涉及的氢吸附和解吸过程同样存在于电解水[10],这一新的发现推动研究人员对磷化钴的电化学性能进行探究,进而开发出新的合成、改性方法并提出了新的应用形式,对磷化钴电化学性能研究的大幕由此拉开。其后的研究表明,磷化钴材料在电化学领域表现出优异的性能,是电化学能源领域极有前景的新型材料。本文总结了磷化钴的物理化学特性、制备方法以及近年来其在电化学能量转化与储存方面的发展现状,提出了存在的问题和可能的发展方向,以期为相关领域的研究提供一定的便利。
1 磷化钴的晶体结构
过渡金属磷化物同时具有金属和陶瓷的特性,键合类型主要为金属键和共价键,通常过渡金属磷化物的富金属组分具有金属特性,富磷组分是半导体且稳定性比富金属组分差[3,11-12]。
对于金属磷化物,一般的通用晶体结构为:金属原子形成三棱柱,非金属原子被包含在中间,对于富含金属的成分,最邻近原子的数量增加,与位于棱镜垂直面中心附近的其他金属原子形成十四面体(TKD)配位,如图1所示[3]。

图1 金属磷化物的晶体结构
磷化钴有多种化学计量,目前CoP/Co2P是热门的研究对象。Co2P 结晶属于Co2Si 结构类型,该结构类型包括边共享的CoP4四面体和CoP5五面体,产生9 配位的P 原子;CoP 按MnP 结构类型结晶,由面共享的CoP6八面体和边共享的PCo6三棱镜组成,这两种结构类型(图2)都可看作是NiAs结构类型的衍生[13-14]。这和上述的金属磷化物的通用晶体结构也是一致的。
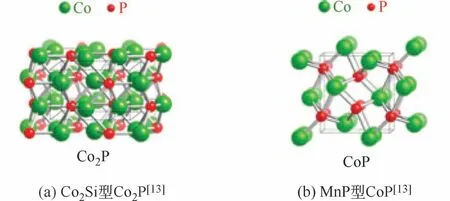
图2 两种化学计量磷化钴的晶体结构
2 磷化钴的制备方法
CoP和Co2P两种物相的磷化钴是目前主要的研究对象。如何设计高效的电化学能量转化与储存的磷化钴催化剂,一直是备受关注的课题。据研究,除了自身的电子特性外,磷化钴的晶体尺寸、形貌和比表面积等对其性能也有重大影响,纳米级别的多孔材料通常表现出更高的催化活性。对于催化加氢和水分解,纳米尺寸的颗粒和丰富的孔结构带来较高比表面积,暴露出更多的活性位点[15-16];对于电极材料,纳米结构电极可通过减少电子/锂离子扩散距离来补偿活性材料中缓慢的电子和锂离子传输,从而实现更高的倍率性能和更高的容量利用率;纳米结构的多孔复合材料颗粒的形成可限制电池充放电期间复合材料活性成分的体积膨胀,进一步改善负极结构稳定性并且极大地稳定固态电解质相界面(SEI)[17-18]。因而,设计和制备纳米尺寸的材料是磷化钴研究的趋势。
制备磷化钴的方法有多种,如化学气相沉积法、程序升温法、有机金属分解法、溶剂热合成法、氢等离子体还原法、电解熔融盐等,各种方法制备的产品性能和形貌等会有所差异,本文对常用的几种方法进行详细介绍。
2.1 程序升温法
程序升温法多用来制备负载型催化剂,先通过浸渍、研磨等手段制备前体,然后在一定气氛条件下(空气、氮气、氩气等)将前体放入设定好升温程序的容器中反应。
Chen 等[6]用Co(NO3)2·6H2O 和NH4H2PO4的混合液浸渍二氧化硅,然后在393K 下干燥12h,823K下煅烧4h,得到Co∶P 为1∶1 的前体。将前体放入反应容器,在流动氢气气氛(以每克前体通入320mL/min 的氢气流量计)下以10K/min 的速度从293K 升温到523K,再以1K/min 的速度从523K 升温到923K,在923K保持3h,得到二氧化硅负载的Co2P、CoP 混合物。这是程序升温法的典型做法。普遍认为在干燥煅烧过程中金属磷酸盐脱水生成金属氧化物和磷氧化物,在接下来的程序升温还原过程中温度应当保持在500℃以上,同时输入氢气(由于磷化物与水快速反应,因此需要适当调高氢气流速以保持磷化物表面水的较低蒸气压[19]),金属氧化物被还原成金属颗粒,磷氧化物被还原成挥发态的P 和PH3,最后磷和磷化氢与磷化金属颗粒生成金属磷化物。根据文献对H2-程序升温还原(TPR)曲线的分析[20-22],推断磷酸盐分解的方程式可能如式(1)~式(3)。

在程序升温法中,初始P/Co 的比值很大程度上决定了生成磷化钴的化学计量。Cecilia等[23]通过保持恒定的钴含量(Co的质量分数为5%)并调节Co(OH)2和H2PO3H 的比例,使初始P/Co 原子比从0变化至2.5,用等量浸渍法负载在商业二氧化硅Cab-osil M-5载体上,将前体在氢气氛围下程序升温至720℃下反应2h得到产品。发现低磷钴比(0.5和1)下生成Co2P 相,高磷钴比(≥1.5)下生成CoP 相。Hu 等[11]对Co 粉和P 粉的混磨产物退火处理,发现200℃时生成了纯CoP相,而大于300℃时出现了Co2P 峰,并且随温度升高越来越明显,可能是温度升高造成了磷损失,这也说明磷相对含量对相形成起到关键作用。
Cecilia 等[4]还制备了一系列负载在不同载体上的磷化钴,发现前体-载体的相互作用对生成磷化钴的化学计量比也有影响。将Co(OH)2和H2PO3H按一定化学计量比反应,得到Co(HPO3H)2溶液。通过等量浸渍法将该成分以钴质量分数10%负载在载体上,然后在100mL/min 氢气气流下,以3℃/min升温到指定还原温度,制备了分别负载在介孔二氧化硅、锆掺杂介孔二氧化硅、商业二氧化硅Cab-osil、γ-Al2O3上的磷化钴。其中在γ-Al2O3上生成Co2P,在其他三种载体上生成CoP。这是因为γ-Al2O3上的前体-载体的相互作用非常强,使还原过程变得困难,可能需要更高的温度才能得到CoP,反而有利于Co2P 相形成。此外,将Co2P 置于高温还原气氛中,同时通入适当的磷源(如PH3),Co2P 可以转化为CoP。
除了负载型催化剂,程序升温法也可用来制备复合材料。例如,Das 等[24]将Co(NO3)2·6H2O、三苯基膦和三聚氰胺混磨,N2保护下以4.7℃/min 升温至850℃退火1h,制得了Co2P 纳米颗粒封装的N、P 双掺杂碳纳米管。Gao 等[25]将多金属氧酸盐和双氰胺在850℃下氮气中退火6h,制备了N 掺杂C 壳包覆的Co2P/WC纳米异质结,在有限空间内同时实现了Co的磷化和W的碳化。
程序升温法虽然操作简便,但是普遍需要较高的反应温度,这可能使小颗粒聚集、微观结构崩塌,生成颗粒大、比表面低的产物,而且高温将导致P 流失,因此常常需要过量的P。传统的程序升温法常把磷源和钴源混合,这使得产物中可能残留磷酸盐等不良导电物质,影响材料整体的导电性,这在电化学应用中会产生巨大的缺陷。
2.2 气相沉积法
为了解决传统程序升温法反应温度高、产物不纯、导电性差的问题,科研工作者对其进行了改良,开发了气相沉积法用于磷化钴材料的制备。典型的做法是在管式炉的上游放一装有次亚磷酸盐的磁舟,隔一段距离在下游放一装有钴盐的磁舟,高温下钴盐在气氛中被还原成钴金属,然后被次亚磷酸盐分解产生的PH3磷化,生成相应的磷化物。
气相沉积法主要起磷化作用,不能改善催化剂整体形貌和性能,构造特殊结构的复合前体成为气相沉积法制备高催化效率催化剂的常用手段。Zhou等[26]将Co(NO3)2·6H2O和聚乙烯吡咯烷酮(PVP)溶解在水中,搅拌至完全干燥,研磨后放在N2中热处理,获得多孔3D碳纳米片封装的Co纳米颗粒前体,接着通过气相沉积合成了基于前体形貌的封装Co2P。Chen 等[27]和Huang 等[28]将钴盐与尿素混溶在水中,在晶化釜中以水热合成得到钴纳米线状前体,接着在管式炉的上游放置一个装有NaH2PO2的磁舟、下游放钴前体,于惰性气氛中在高温下磷化,合成了Co2P纳米线阵列。
由于严苛的反应环境,不管是电解水还是电极都对材料结构的稳定性和比表面积提出一定要求,而其中MOFs材料因其优异的稳定性和较高的比表面积备受关注。MOFs 材料是一种制备多组分微纳米复合材料的理想前体,衍生自MOFs前体的复合材料具有丰富的孔结构,有利于暴露更多的活性位点,有机配体形成的碳骨架可以提高导电性和结构稳定性,保护活性组分。Jin 等[29]将Co(NO3)2·6H2O和2-甲基咪唑溶解在甲醇中,构筑了正十二面体的钴基金属有机骨架ZIF-67,经过退火、磷化,再于高压釜中与氧化石墨烯复合,成功合成了继承前体形貌的石墨烯复合Co2P 颗粒。通过类似的方法还制备了生长在多孔碳多面体上的N掺杂碳纳米管中镶嵌的Co2P[30]、包覆在碳骨架里的钴基双金属磷化物[31]、镶嵌在泡沫镍支撑的石墨烯上的CoP/碳多面体核壳结构[32]等。
由于气相沉积法中磷源多使用次磷酸盐或次亚磷酸盐,大大地降低了反应温度;同时,磷源和钴源并未混在一起,磷酸盐类非活性物质也就不会混杂在产物中,有利于制备纯度高的产物。另外,钴或氧化钴前体的形貌或分散度等性质一般会保留至最终合成的磷化钴材料中,有利于合成高分散度或特定形貌的磷化钴材料。然而反应时产生剧毒易爆炸的PH3,操作过程需要特别注意安全问题。
2.3 有机金属分解法
有机金属分解法通常用金属有机物作为钴源,一般先需要热解生成钴纳米粒子,然后进一步磷化。常用的钴源有八羰基二钴、乙酰丙酮钴等,常用的磷源有三辛基膦和三苯基膦等。
Callejas 等[13]采用钴纳米颗粒制备和钴纳米颗粒磷化两步制备了Co2P和CoP。首先在混合有机溶液中热解八羰基二钴得到钴纳米颗粒,热解过程中通Ar 气保护。接着将制备的钴纳米颗粒注入经过除杂质、氩气保护下升温到290℃的1-十八烯和三辛基膦混合物中保持40min,得到尺寸均一的空心Co2P纳米颗粒;而在330℃的1-十八烯、油胺和三辛基膦混合物中保持60min,得到空心CoP 纳米颗粒。采用类似的合成方法,Pan 等[33]调整磷源合成了不同的磷化钴催化剂,使用三辛基膦合成CoP基催化剂或以三苯基膦合成Co2P基催化剂。
通过向溶剂里添加特定组分还可以对产物进行改性,例如,Xie 等[34]预先将石墨烯超声混合在油胺溶液中,合成了石墨烯修饰的Co2P-Co空心纳米球。Chen等[35]发现通过控制升温速率,可以合成不同形貌的磷化钴。以乙酰丙酮钴作为钴源,将其溶解在油胺中,在氮气鼓泡和磁力搅拌下升温至100℃保持1h。随后加入三苯基膦到溶液中保持1h,迅速加热至300℃保持3h,根据不同的升温速率会得到不同形貌的产品,其中升温时间为20min时得到Co2P纳米棒产物,升温时间为25min时得到Co2P 纳米花产物。Cheng 等[36]则将乙酰丙酮钴溶解在油胺和甲苯的混合液中,与加入的三苯基膦鼓泡搅拌,接着迅速升温至280℃保持2h,合成了中空直径约110nm的Co2P纳米花。Hou等[37]以三苯基膦作表面活性剂,在油胺溶液中还原了直径约6nm的Ag颗粒,接着加入乙酸钴在Ag颗粒表面成壳,被磷化成CoxP,形成了Ag@CoxP核壳结构。
表1总结了通过有机金属分解法合成磷化钴的条件。
目前很少有关于磷化钴生成机理的报道,不同化学计量的磷化钴相互转化的条件也尚不明确,但通过表1可以粗略了解到,在有机金属分解法中生成CoP比生成Co2P往往需要更高的反应温度和更长的反应时间,这使CoP的结晶度更高,另外通过添加三辛胺、甲苯等辅助剂,可以合成特殊的磷化钴形貌。有机金属分解成的金属单质非常活泼,因此反应全程需要惰性气体保护,注入的溶剂通常需要脱气处理,操作相对繁琐。但该方法易于合成多种特定形貌的磷化钴纳米材料,可能对于一些利用形貌效应而提高应用性能的体系具有一定的意义。

表1 有机金属分解法合成多种形貌磷化钴的条件总结
2.4 溶剂热法
在溶剂热法中溶剂扮演了一个重要角色,通过添加不同种类的溶剂可以调控前体的形貌,从而调控最终产品的形貌。
Huang 等[39]将磨成微米级的红磷加入到十六烷基三甲基溴化铵(CTAB)和Co(NO3)2·6H2O的混合溶液中,装入反应釜在200℃反应48h,一锅法制备了厚度3~11nm 的Co2P 纳米片。Liang 等[40]将钴盐和黄磷溶解在CTAB的水溶液中,只需要在微波辅助下处理10~30min就可以获得Co2P纳米梭。由于溶剂高温蒸汽压或高温分解的限制,对于反应温度要求不能太高,因而该类合成方法所使用的磷源大多为单质磷,从而使反应温度在200℃左右,但高压的反应环境对于设备材质要求很高,且具有一定的危险性。
2.5 其他合成方法
Guan 等[19]用氢等离子体还原法制备了磷化钴。用(NH4)2HPO4调节Co(NO3)2·6H2O 溶液的pH,形成的沉淀经干燥、焙烧、破碎、筛选后得到氧化钴前体,然后放入反应器磷化,在90V下反应60min得到纯的CoP晶体。该方法所需温度不高,不会造成大量的P 损失,因此不需要过量的P,反应过程中不会产生PH3。
Jiang等[41]采用超分子凝胶辅助法制备了锚定在多元素共掺杂石墨烯上的Co2P 颗粒。先将三聚氰胺、Co(NO3)2·6H2O和H3PO4混合均匀,加入自制的氧化石墨烯,搅拌后形成凝胶,接着冷冻干燥,在Ar气氛中900℃热解2h,获得产品。
酵母菌表面带有丰富的负电荷,可以吸附金属离子。Li等[42]利用这个特点制备了多级孔的磷化钴纳米材料。酵母菌自带磷源,带负电荷的表面会吸收过量带正电的钴离子,然后带正电的酵母菌与带负电的氧化石墨烯牢牢吸附。随后用水热法初步碳化酵母菌,形成Co3O4/碳化酵母菌/还原氧化石墨烯(rGO),最后再在高温和混合气氛下煅烧,得到Co-Co2P@N、P 掺杂碳基质/rGO 材料,其中Co-Co2P 纳米颗粒的直径约104.7nm。作为磷源和模板,酵母菌价格低廉,比表面积高,反应过程中不会产生有毒的PH3。
基于盐模板与目标晶体晶格匹配理论,Li等[43]制备了暴露特定晶面的2D 单晶Co2P。(NH4)2HPO4溶于乙醇、KCl 和去离子水的混合溶液作为P 源,Co(NO3)2·6H2O 溶于乙醇作为钴源,依次均匀涂覆在KCl模板表面,高温下气氛中退火,得到了包覆在盐模板上厚度约5nm 的Co2P 层。盐模板可以方便地通过水洗去除。
3 磷化钴的应用
3.1 作为电解水催化剂
众所周知,电解水是最理想的清洁能源技术之一,如果能开发出绿色高效的电解水催化剂,应用在燃料电池和太阳能电解槽等清洁能源技术中,全球能源危机和环境危机将得到极大改善[10,44]。电解水可分为析氢反应(HER)和析氧反应(OER)两个半反应,目前性能最优异的商业HER 催化剂是Pt/C,最优异的商业OER 催化剂是RuO2和IrO2,然而高昂的价格和稀少的储量严重限制了其工业化应用[45-46]。过渡金属磷化物(TMPs)中引入的P原子使费米能级附近的态密度得到提升,能以较小的超电势和快速的反应动力学来触发质子还原,具有与贵金属相似的特性,因此将TMPs 应用在电催化方面是有可能的[10]。2005 年Ping 等[47]首次预测Ni2P 的(001)面是HER的高活性位点,2013 年Lewis 等[48]采用溶剂热法制备了一种(001)晶面暴露密度极高的Ni2P 空心微球用于水裂解,发现它是活性最好的非贵金属HER催化剂,证实了预测的有效性。磷化钴中Co 和P分别带少量正电和负电,带电性质类似于Ni2P中的氢化物受体和质子受体,导致相似的催化机理,这使得磷化钴也具有高HER 活性[49-50]。Sun等[2]总结了TMPs 催化剂上整体析氢的机制[式(4)],并认为H2的产生可能包括三个单独的步骤,如式(5)~式(7)。

式(5)被称为Volmer 反应,当作为阴极催化剂时,TMPs 首先会在一个较低的过电位下还原质子,生成的H 吸附在催化剂表面。接着通过以下任一步骤生成H2:当一个自由的H+与一个吸附的H 原子反应生成H2,称为Heyrovsky 反应[式(6)];当两个吸附H 原子结合生成H2,称为Tafel 反应[式(7)]。典型的HER 过程都遵守Volmer-Heyrovsky机理[51]。
Callejas 等[13]用有机金属分解法成功制备了尺寸均一的空心Co2P 和CoP 纳米颗粒,并将产物以1mg/cm2涂布在Ti箔上,来对比二者的HER催化性能。发现在0.5mol/L H2SO4中要产生-10mA/cm2和-20mA/cm2的阴极电流密度,CoP/Ti分别需要-(75±2)mV和-(90±3)mV的过电势,在相同条件下进行测试的Co2P/Ti电极,所需的过电势分别为-(95±3)mV和-(109±4)mV。进一步通过加速降解研究和恒电流测试,发现在+5mV 到-140mV 电压范围内循环500圈后,CoP/Ti电极达到-20mA/cm2的过电压仅增长2mV而Co2P/Ti电极增长了10mV,如图3(a)所示。在0.5mol/L H2SO4中恒电流测试24h后,CoP/Ti电极达到-20mA/cm2的过电压增长11mV 而Co2P/Ti 电极增长了24mV,这说明CoP/Ti 的稳定性也明显优于Co2P/Ti。二者形态等效,平均Co—P 键长基本相同,据此推断磷化钴的结构和键合不会显著影响HER催化活性,但是相同条件下CoP的活性始终比Co2P高。Li课题组[14]的工作解释了这一点,经过离散傅里叶变换(DFT)计算发现,磷化钴(211)晶面上暴露的Co 原子才是真正的H 吸附的活性位点,CoP 和Co2P(211)晶面上有4 种可形成Co—H 键的H*吸附构型,分别标为(a)、(b)、(c)、(d),如图3(b)所示。CoP的(211)晶面上的所有“a-c”单Co 位点的绝对吸附自由能都在0.1eV 以内,远低于Co2P(211)面上相应位点,Co2P 对H*的强吸附削弱了HER 活性,而CoP 高的P 含量可以缓解这种强吸附。在OER 反应中Co2P 的活性高于CoP,这是因为Co2P 表面暴露了更多的Co,可以形成丰富的OER 活性物质Co2P@CoOOH 异质结[52]。多质子偶联的电子转移步骤导致OER 的反应动力学非常缓慢,需要活性高的催化剂来克服水解过程中较高的活化能垒,因此在很大程度上OER 成为电解水的控制步骤[37,53]。
然而纯的磷化钴往往面临着比表面积低、暴露的活性位点不足、导电性低、易受电解液腐蚀等问题。为了克服Co2P 上强吸附作用的缺陷,Gao 等[25]以一种多金属氧酸盐作钴源和钨源,与双氰胺一起退火,合成了碳包覆的Co2P/WC异质结。过渡金属W 具有和Pt 类似的d 带电子结构,生成的WC(001)和Co2P(001)耦合成均一的杂化体,可以得到一个非常弱的H*吸附自由能(-0.17eV),而当与掺杂了吡咯N的碳材料复合后,H*吸附自由能进一步降低到-0.1eV,有利于HER 活性。除此之外,多层包覆的碳材料还可以防止活性物质团聚,保护其不受酸性电解液腐蚀,材料导电性也得到了明显提高,电荷转移电阻仅11.43Ω,在0.5mol/L H2SO4中只需要91mV 过电压就可达到10mA/cm2电流密度。Guo 等[54]采用原位一步自组装和密闭热解法制备了N 掺杂碳纳米管包覆的Co2P-CoN 双活性中心催化剂,Co2P-CoN 被封装在碳纳米管中,拥有单个孤对电子的吡咯N既均匀分布在碳纳米管中又存在于活性颗粒内部,被认为是活性催化中心,如图3(c)。1D 碳纳米管可以提供有序通道以实现快速物质传输和电子转移,且与Co2P-CoN 的强耦合作用大大提高了导电性。生物质模板同样被用于改善磷化钴催化剂,Li 等[42]利用酵母菌自身电荷特性均匀地吸附钴离子,然后与氧化石墨烯复合,在高温下退火得到了生长在碳化酵母菌表面的Co-Co2P 纳米颗粒。酵母菌本身具有高比表面积,生长在碳化酵母菌表面的Co-Co2P 纳米颗粒高度分散,能提供更大的电化学催化面积。如图3(d),Co-Co2P@NPC/rGO 的Rs只有2.09Ω,低于碳棒,意味着具有良好的电导率,Co 单质的贡献不可忽视。再加上杂原子的协同作用,该材料在0.5mol/L H2SO4中只需61.5mV过电势就可达到10mA/cm2的电流密度,并且可以在1000mA/cm2的电流密度下维持20h,这是因为碳结构提供了良好的保护。这里杂原子对HER性能的提升同样是通过调控电子结构来优化氢吸附自由能实现的[33,51]。Hou等[37]也试图通过引入金属单质来提升电导率,以三苯基膦为磷源,在含Ag 颗粒的有机溶液中热分解乙酸钴,构筑了Ag@CoxP核壳结构,阻抗图表明该材料的电荷转移电阻仅2Ω,低于Co2P(3.5Ω)。Ag纳米颗粒还可以调控CoxP壳的电子结构并增强CoxP的活性。在OER反应中,CoxP部分转化为CoOx,这被认为是OER反应的活性中心。Dutta 等[55]将钴盐溶解在烷基胺中,加热到230℃后通入PH3气体,再经过退火得到针状窄六方的Co2P 纳米颗粒。作为OER 催化剂,在1mol/L KOH 中310mV 过电压下即可达到10mA/cm2电流密度,甚至优于基准的IrO2/C催化剂,这归因于部分氧化的表面物种使载流子传输更加方便和窄针状结构带来的较大活性比表面积。

图3 文献所制备用于电解水的磷化钴的各项性能曲线和形貌图

表2 各种磷化钴电解水催化剂达到相应电流密度所需过电势汇总
在碱性环境下的HER 需要的过电势高于酸性环境,这是因为水缔合步骤增强了能量壁垒。就实际应用而言,设计具有HER 和OER 双活性位点并且在碱性条件下能表现出高全水解活性的催化剂仍是一个巨大挑战。鉴于金属磷化物表面氧化有利于OER 反应,而混合金属磷化物可调节H*吸附自由能,Duan 等[45,57-59]以三苯基膦作磷源,通过退火生长在泡沫镍上的钴铁氢氧化物,制备了Fe、O掺杂的Co2P作为全水解催化剂,其中O取代了Co2P晶胞中的部分P原子,Fe取代了部分Co原子,形成组分均一的复合物,当用于全水解反应达到10mA/cm2时所需过电势低至335.5mV, 优于Pt/C-IrO2(491.5mV),这归因于Fe/Co和P/O掺杂比例的合理调节和三维泡沫镍基底对电导率和电子转移的提升作用。Li等[56]用尿素辅助沉淀法制备了CoAl水滑石前体,然后经过磷化合成了分层的花状CoP/CoP2/Al2O3复合物。分层纳米结构可以提供大比表面积和活性位点,以实现快速的电子/离子传输路径,在1mol/L KOH 中分别需要300mV(对OER)和138mV(对HER)来达到10mA/cm2的电流密度,用于全水解时只需要1.65V就可以达到10mA/cm2的电流密度。各种磷化钴电解水催化剂达到相应的电流密度所需过电势见表2。
多金属协同作用和高分散度带来的较高活性表面积,可以取得良好的催化性能,但并不能解决在电化学过程中必然会出现的活性成分聚集和剥离,进而影响材料长期寿命的问题,目前大多同时采取分隔限位的复合手段。有研究者采用一种直接热解法制备了碳纳米片/纳米管封装的Co2P颗粒,超细的Co2P纳米颗粒均匀地嵌在多孔纳米片中,有效防止了聚集和剥离,材料整体的比表面积高达199.94m2/g,可以提供大量的活性位点。杂原子和碳材料的协同作用提高了材料的电导率,制作为碱性电解槽时,在1mol/L KOH中提供1.64V过电势即可得到10mA/cm2的电流密度,并且在该条件下工作25h 后仅损失2.1%的电流密度,表现出优异的稳定性[60]。Song等[31]合成了碳骨架封装的Cu-Co双金属磷化物,通过调节两种金属的比例,发现Cu0.3Co2.7P/NC性能最优异。Cu 被均质取代进入CoPx中,起到了提高电导率、降低氢吸附自由能的作用。在1mol/L KOH中用作OER 电极时,达到10mA/cm2只需要0.19V,优于商用RuO2(0.27V),这是因为Cu0.3Co2.7P/NC有高的电化学活性面积和相对低的电荷转移电阻[图3(e)和图3(f)]。特别是作为阴阳极组装成电解槽后,达到70mA/cm2电流密度仅需过电势1.74V,工作50h 后活性几乎没有损失,这是因为其自身结构的稳固和碳骨架的保护作用。不管是封装还是包覆,都是在活性材料之间、活性材料和电解液之间作出一定的限制和分隔,稳定的结构保证长时间的寿命,这已经得到了多个文献的证实。同时,目前报道的封装或包覆大多使用碳材料与活性物复合。碳材料本身具有高导电性,有利于电荷转移;高结构稳定性和耐受性,可以维持稳定的结构防止活性物质聚集和腐蚀;丰富的微孔结构提供高活性面积[61-62]。另外,碳的来源丰富,处理后的各种碳源可以提供杂原子,如N、S、P 等,通过调节电子结构和表面极性来构造碳缺陷从而产生活性位点,并且优化氢的吸附自由能,进而提高复合材料的催化活性[51,63-64]。目前改性磷化钴材料的催化性能已经接近相应的贵金属基准物,但活性位点生成和作用机理仍不够明确,需要进一步探究。在酸或碱性电解液的环境中,电极往往会腐蚀,电极结构受到破坏,因此需要开发具有更强耐受性的磷化钴材料或者提供增强的保护作用。另外该合成方法往往复杂且条件苛刻,不适用于大规模生产,因此优化合成工艺并应用于实际生产也是下一步研究的方向。
3.2 用于超级电容器
超级电容器具有功率密度高、充放电快、寿命长等优点,在混合动力汽车、可再生能源发电厂、大型工业设备等方面应用广泛[2,65]。根据储能机理,超级电容器可分为两种:电双层电容器通过在电极表面吸附静电电荷储存能量;赝电容器依靠电极材料表面或者表面附近发生可逆氧化还原产生法拉第准电容实现能量的转化和储存。然而,电双层电容器比容量和能量密度低,赝电容器功率密度低、循环稳定性差,不能满足人们对高性能电容器的要求,设计由性能优异正负电极组成的不对称超级电容器(ASC)来扩大电压窗口,是一种行之有效的提升方法[66-69]。通常碳材料作为超级电容器负极,金属氧化物、金属氢氧化物、金属硫化物和聚合物等用作正极,然而这些正极材料导电性普遍较低,导致高功率密度下的快速电子传输动力学缓慢[10-11]。而作为TMPs 的一种,磷化钴既有准金属特性又有良好导电性,在磷化钴中同时存在着还原的钴和氧化的钴,还原的钴可以提供自由电子并增加导电率,氧化的钴可以通过法拉第反应储存电子提供高电容。近年来,作为优异的超级电容器电极材料受到广泛研究[2-3,70]。
在保持高功率密度的前提下,提高超级电容器的能量密度,是改良超级电容器性能的大方向,而制备具有特殊形貌的纳米材料是常用的提升策略。目前可以通过两步法有效地获得特殊形貌的磷化钴,首先采用尿素辅助制备可控形貌的磷化钴前体,接着在管式炉内经气相沉积法低温磷化。基于两步法,3D分层绒球状CoP空心微球[71]、生长在碳布上的CoP纳米线阵列[72-73]等被合成出来,这些3D形貌的材料本身具有大的比表面积,提供大的界面接触面积和活性位点,纳米尺寸使电子/离子传输路径变短,因此表现出优异的电化学性能。有机金属热分解同样可以构筑特殊形貌的磷化钴。Chen等[35]以乙酰丙酮钴作钴盐、三苯基膦作磷源,在热的有机溶剂中合成了Co2P纳米花和纳米棒。巧妙的是,纳米花和纳米棒的形成完全是通过差异化磷化过程的升温速率实现的,升温20min得到纳米棒,升温25min 得到纳米花[图4(a)和图4(b)]。将Co2P 纳米棒和纳米花用作超级电容器电极材料,设定电流密度为1A/g,分别得到284F/g 和416F/g 的比容量。此外,以Co2P纳米花作阳极、石墨烯作阴极,组装了不对称超级电容器,分析其阻抗图发现等效串联电阻为0.87Ω,表明ASC良好的导电性和低的内阻[见图4(c)],在0.3kW/kg 的功率密度下显示24W·h/kg的能量密度,并且在6000次循环后仍可保持97%的比容量。通过引入甲苯和油胺的混合溶液作溶剂,Cheng 等[36]同样用有机金属分解法在类似条件下制备了中空的Co2P纳米花,直径110nm左右,表层包覆着薄的碳壳,电荷转移电阻Rct和内部电阻Rs分别只有0.17Ω和0.64Ω,表现出良好的快速电子、离子转移能力。将其用作超级电容器电极材料,在1A/g电流密度下可提供412.7F/g 比容量,用于不对称超级电容器时,在850W/kg 功率密度下表现出30.5W·h/kg 能量密度,在5A/g 下经过10000 次循环,比容量保持率高达108.0%[图4(d)],即使功率密度增加到8500W/kg,输出的能量密度也可达22.8W·h/kg,表现出优异的循环稳定性和倍率性能。
研究者们还对掺杂元素的作用进行了探讨。例如,Lan等[74]报道了基于泡沫镍合成的分层的Mn掺杂CoP纳米线修饰的纳米片簇阵列,得益于独特的双金属纳米结构和协同作用,该材料拥有比未掺杂材料更高的面积比电容,用作不对称超级电容器负极时在193W/kg 和1939W/kg 处分别有35.21W·h/kg和30.87W·h/kg的能量密度。Elshahawy 等[75]将碳布上生长的CoP 纳米管阵列与硫粉分置管式炉两端,退火后合成了S 掺杂的CoP 纳米线阵列,S 掺杂后P/P-O 的比例由20%提升至40%[图4(e)],证明CoP表面的钝化层减少,整体电导率提高了并促进了电子转移。分析循环后的S掺杂CoP的化学态,发现Co2p光谱的位移小于纯的CoP,表明S的引入也改善了循环稳定性。组装成混合超级电容器后,在0.8kW/kg 功率密度下能量密度可达39W·h/kg,并且在循环50000次后保留初始容量的86.4%。
磷化钴应用于超级电容器,目前主要集中于通过多孔隙的复杂形貌(分层结构、纳米棒、纳米花等)的构筑来获得大比表面积、暴露更多氧化还原反应位点的研究。另外,为了提高导电性,除了与碳基材料复合,也有通过非金属元素(如S)或金属元素(如Mn、Ni[76]等)掺杂来提升性能的研究报道。然而对于催化活性位点的形成、催化机理仍缺乏深入了解,需要未来通过多种测试手段联合分析,另外由于对多孔隙、高比表面积的追求,材料的体积能量密度往往不够理想,应该重新审视材料尺寸和形貌设计方向。各种磷化钴材料用于超级电容器性能数据见表3。

图4 文献所制备用于超级电容器的磷化钴的各项性能曲线和形貌图

表3 各种磷化钴材料用于超级电容器性能数据汇总
3.3 新型电池
锂离子电池不仅环境友好,而且具有能量密度相对较高、质量轻、体积小、循环寿命长、无记忆效应等优点,在电动汽车、便携式电子产品的能量储存等方面颇具潜力,近年来受到大量关注[77-78]。锂离子电池的作用依赖于Li+的可逆穿梭和正负极之间的电化学差异,因此电极材料的本征特性极大程度地决定了电池的整体性能,而传统的锂离子电池电极材料,例如已经商业化的负极材料石墨电极,理论容量仅为372mA·h/g,已无法满足日益增长的能源需求[79]。
为了实现更高能量密度、功率密度以及快速充放电的需求,研究者们对基于转化反应的金属硫化物、氧化物、氟化物和磷化物等电极材料进行了大量研究。过渡金属磷化物(约0.4V)的优势在于平均极化率低于硫化物(约0.7V)、氧化物(约0.9V)和氟化物(约1.1V),这意味着电化学反应动力学良好,而且P基负极拥有合适的电位,可防止锂枝晶形成并在组装成全电池时提供高电压,自身的金属特性也会带来良好的导电能力,因此是非常有潜力的锂离子电池负极材料,而磷化钴是其中性能较优异的代表[80-81]。2002年,Nazar等[82]将CoP3应用于Swagelok 型电池以探究该材料吸收锂的特性,研究发现整个过程涉及一个新颖的机理;CoP3吸收锂会转化为Li3P,高分散的钴簇镶嵌在Li3P基质上。后来的研究者们又把该机理推广到过渡金属磷化物中,即作为电极的情况下,通常MPy会转化为镶嵌在Li3P基质中的金属纳米颗粒,形成复合电极,转化机制如式(8)[18]。

相比其他电极,这种转化反应形成的复合电极提供更高的锂离子交换效率,有利于产生更高的理论容量[17,83-84]。据报道,基于可逆转化反应的磷化钴具有很高的理论比容量(CoP 理论比容量894mA·h/g,Co2P 理论比容量540mA·h/g),被认为是一类非常有应用前景的锂离子电池负极材料[85-87]。
近年来,文献报道的磷化钴材料的比容量已经明显高于商业石墨电极。例如,Kwon 等[18]通过固相合成路线,合成了碳改性部分歧化的磷化钴二元产物(CoP 和Co2P,CoP 是活性组分而Co2P 是惰性组分,在电池内Co2P 起着减轻体积变化的作用),拥有高达531mA·h/g 的初始容量,200 个循环后仍可保持为407mA·h/g。尽管如此,离理论容量仍存在差距,循环过程中容量衰减问题依然明显。造成容量衰减的原因有很多:一是电化学反应过程中活性颗粒聚集,暴露的活性面积减少;二是在充放电循环中,锂化/去锂化会引起巨大的体积变化,活性物质破碎并且与集流体失去接触;三是电解质的分解和固体电解质中间相(SEI)的形成会导致部分不可逆的容量损失。对于该问题,常采用的解决方法有两种:一是合理设计磷化钴的微观结构,如纳米球、纳米棒、中空球、核壳结构等,可以有效改善电解质和电极之间的界面接触面积并适应体积变化;其次为活性物质提供适宜的结构保护,既可增强导电性又可以缓解体积效应,而在实际研究中通常将两种方案结合使用[85,88-90]。
将磷化钴与功能碳材料复合是一种有效的方式,这是因为功能碳材料普遍具有以下优点:首先碳材料本身具有导电性,可以增强电子传输,提高材料的电导率;其次碳材料可以构建复杂的形貌,被赋予大量孔道,从而起到阻止活性物质聚集、提供活性表面、加快电子传输和质量传输、缓解体积变化的作用[91-92]。石墨烯是近年非常热门的复合材料。Lu等[87]用油酸和三辛胺做辅助剂,在石墨烯纳米片上锚定了海胆状的Co2P,复合后的产物具有增强的导电性,用于锂离子电池负极,当电流密度分别 为200mA/g、400mA/g、800mA/g、1200mA/g、1600mA/g 时,分别获得521mA·h/g、419mA·h/g、361mA·h/g、330mA·h/g 和312mA·h/g 的比容量,电流恢复到200mA/g 时容量仍保持在520mA·h/g,远远优于未复合的Co2P,并且随循环次数增加比容量也获得提升,这是因为循环过程中海胆状Co2P破碎,更好地分散在石墨烯纳米片上且受到了石墨烯结构限制而不至于脱落,增加了活性表面积。Xie 等[34]设计了石墨烯复合的Co2P-Co 空心结构,空心的Co2P-Co纳米颗粒均匀分布在石墨烯上。Co金属颗粒的存在提高了电导率,同时可以作为催化剂促进SEI分解,所以在后续的循环中表现出上升的比容量。石墨烯复合化引入了大量中孔,提高了比表面积的同时也可稳定材料的整体结构,更重要的是Co2P-Co纳米颗粒的中空结构和纳米颗粒与石墨烯之间的空间可以为体积波动提供缓冲空间,这保证了电池的循环稳定性,在100mA/g电流密度下循环200次仍保有929mA·h/g的可逆容量。以Cu2O纳米立方体作模板,石墨烯包裹的CoP纳米笼也被合成出来,如图5。CoP 纳米笼具有强大的结构稳定性和对体积变化适应能力,通过冷冻干燥技术引入的石墨烯具有高孔隙率,可提高整体电导率和电子传输效率,在500mA/g 高电流密度下500次循环后容量保持在460.4mA·h/g,这与材料结构的稳定性密不可分[93]。为了使活性组分与碳基质的结合更稳固,防止活性材料聚集和脱落,Wang 等[94]合成了一种多维分层碳材料限位的磷化钴纳米颗粒,通过两次水热反应将CoP层层限位,CoP先被分散在多孔碳壳中,碳壳表面再包覆一层厚约10nm 的碳壁,最后整体嵌入到石墨烯纳米片组成的3D 网络中,如图6。得益于三层碳材料提高的导电性能和多重框架保护,在0.25A/g电流密度下有754mA·h/g的比容量。除此之外,生长在石墨烯上的分层Co2P微球[95]、生长在石墨烯上的CoP 纳米线[96]、嵌入到石墨烯纳米片网络的CoP[97]等均被设计合成,并且都展现不俗的电化学性能。
石墨烯复合物固然拥有非常优异的电化学性能,然而高昂的成本极大限制了其应用,其他相对价廉易得的复合碳材料也可以获得类似的性能。MOFs材料是一种制备多组分微纳米复合材料的理想前体,衍生自MOFs 前体的复合材料具有丰富的孔结构,有利于电子和质量传输、暴露更大的活性面积同时容纳体积变化,稳固的碳骨架可以保障整体结构稳定性并且改善电导率。Lei等[30]对钴基前体ZIF-67进行热解、磷化,制备了具有空间限制的Co2P复合材料。材料的主体是多孔的碳十二面体纳米材料,表面有大量碳纳米管缠绕,Co2P颗粒镶嵌在碳纳米管内部,既缓解了体积膨胀,又增加了负极电导率,在100mA/g 电流密度下循环300 次,容量稳定在857mA·h/g,库仑效率高达99.4%,甚至在6.4A/g高电流密度下还可保持508mA·h/g的比容量,表现出良好的倍率性能,如图7。Liu等[99]报道了镶嵌在N掺杂碳基质里的CoP纳米颗粒,碳基质来源于热处理的ZIF-67,CoP均匀地分布在碳基质中,而N掺杂可以提高活性材料和锂离子接触和框架的稳定性。Zhu 等[98]添加碳纳米管合成了改性的ZIF-67 前体,如图8,碳纳米管贯通ZIF-67前体并缠绕成碳网络,提高了电极内电子和离子的传输效率。磷化后这样的结构并未遭到破坏,生成的CoP均匀分布在碳骨架中,可以极好地适应充放电过程中的体积变化,N/P掺杂则改善了电极的电导率,该电极在5A/g电流密度下的比容量高达533mA·h/g,表现出极优异的倍率性能。研究者们还对温度的影响进行了探讨,将红磷和ZIF-67混合物退火,发现700℃形成纯CoP相,800℃形成CoP 和Co2P 复合相,900℃形成纯Co2P相,并且复合相表现出最出色的容量和长时间寿命(0.1A/g下循环100C后1224mA·h/g,1A/g下循环1800C后仍保持在400mA·h/g)[100]。

图5 石墨烯包裹的立方体CoP纳米笼的合成简图[93]
由于锂的储量相对有限,而钠价廉易得、储量丰富,在电解池的氧化还原反应中,钠离子表现出与锂离子相同的嵌入和脱嵌行为,因此近来钠离子电池作为锂离子电池的替代产品,开始受到广泛关注[2,101]。然而钠离子的直径高于锂离子,这导致在钠离子电池系统中电子和质量传输动力学缓慢,钠化/去钠化过程中体积波动较大,因此通常表现出比锂离子电池差的电化学性能[102-103]。与锂离子电池类似,构建具有缓解体积变化的结构可以较好地改善这个问题。Zhou 等[26]以聚乙烯吡咯烷酮为碳源,利用硝酸钴热分解释放气体的吹塑作用,制备了由封装了Co2P 纳米颗粒的碳纳米片组成的多孔3D 碳纳米片网,弹性的碳结构可良好地适应体积变化,大量孔道可提供足够的活性位点和快速的电子传输、质量传输,50mA/g循环100次之后比容量保持在306mA·h/g。Jin 等[29]制备了十二面体形的N掺杂碳基质复合的Co2P 纳米颗粒,材料的形貌继承自相应的MOFs前体,Co2P颗粒被多孔碳和石墨烯基质包覆,如图9,既增强了整体导电性又缓解了Na3P 形成导致的体积膨胀。Zhang 等[104]将MOFs前体和红磷混磨,煅烧后CoP活性成分被包覆在碳纳米片中,而且碳纳米片和CoP 中存在着的P—C键使二者接触紧密而牢固,碳纳米片增强了电导率并适应体积膨胀带来的应力,最终在1A/g 电流密度下循环900C 后仍可将比容量保持在386mA·h/g。这里也提出了CoP的反应机制。在第一次放电过程CoP与钠离子反应生成Co和Na3P,如果CoP粒径足够小而且被限制在导电基质中,那么在第一次充电中优先发生转化反应[式(9)],如果CoP 粒径大且形貌分布不规则,并未包裹在导电基质中,则优先发生去合金反应[式(10)]。如果既有限制在导电基质内的小CoP 颗粒,又有游离在导电基质外的大颗粒,那么两种反应同时发生。

图6 Wang等合成的多维分级碳材料限位的CoP[94]
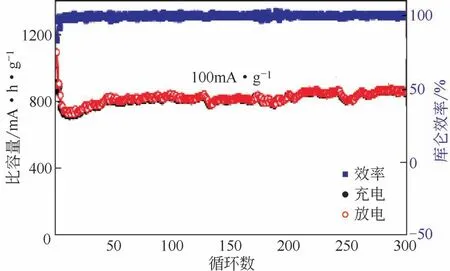
图7 Lei制备的磷化钴的碳复合材料在100mA/g下的循环表现[30]

图8 Zhu等制备的CNTs改性的ZIF-67前体的TEM图[98]
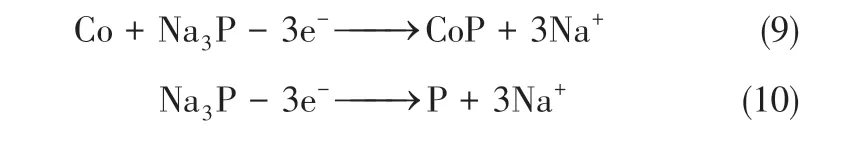

图9 Jin制备的Co2P@N-C@rGO的SEM图[29]
以普鲁士蓝为模板石墨烯改性的双金属磷化物核壳结构也被设计出来,多孔的FeP 作为核被CoP层包裹成立方体,而石墨烯连接所有立方体,形成了分层多孔结构,材料整体同时具有丰富的孔道、普鲁士蓝形成的碳层和CoP外壳三种手段应对体积变化,保障了循环稳定性[105]。此外,非晶态的Co2P被认为可以容纳体积膨胀,也是高容量保持率的电极材料[106]。Ge 等[32]用生长在泡沫镍上的氧化石墨烯改性ZIF-67前体,构筑了3D多孔的石墨烯网络,CoP被封装在有空间余裕的碳多面体内,该材料的优点为不需要黏结剂和导电剂,就能发挥出毫不逊色的性能。
作为锂/钠离子电池电极,磷化钴材料在转化反应过程中存在巨大的体积变化,导致非常差的循环性能,很多研究者将解决问题的希望寄托于合成纳米结构的材料。各种磷化钴复合纳米材料用于锂/钠离子电池的性能表现见表4。事实上,对于近年报道的性能优异的磷化钴材料,无论合成了怎样的形貌,最终活性组分磷化钴的特征尺寸普遍维持在纳米级,这基于纳米级材料可以承受更大的应力而不会发生结构破坏,另外纳米材料中短的离子和电子传输路径使得扩散速率加快,暴露的高表面积有利于高的Li+/Na+通量穿过电解质/电解液界面,提供良好的速率能力[107-108]。通过有机钴源、有机磷源、金属-有机骨架等多种方式引入碳材料,以将活性物质限制在一定空间范围内或者直接在表面包覆一层结构稳定的外壳,也能起到缓解体积变化的效果,并且可以提高材料的导电性能。但另一个问题随之而来,高分散高孔隙带来巨大的活性表面积,使得形成SEI 膜的过程和电极-电解质表面副反应不可逆地消耗大量Li+/Na+,因此初始库仑效率往往非常低。在装成电池前向电极材料提供补偿的Li+可以有效提升初始库仑效率,即预锂化。Zhang等[109]将Li金属溶解在联苯-四氢呋喃溶液中制备了预锂化试剂,在组装电池前将电极材料浸渍30s,发现半电池的初始库仑效率可达到惊人的106%。Forney等[110]将稳定的锂金属粉末制成甲苯悬浮液滴在电极上,结果第一个循环不可逆容量损失降低了20%~40%。如何补偿第一个循环中不可逆的离子消耗,是磷化钴材料迈向实际应用的重要一步,也是未来研究的方向之一。

表4 各种磷化钴复合纳米材料及其用于锂/钠离子电池性能表现汇总
4 结语与展望
本文概述了磷化钴材料的物理结构、合成方法、生效机理等以及近年来分别应用于电解水、超级电容器和锂/钠离子电池等领域的最新进展。
在电解水反应中,活性材料有时会处于苛刻的电解液环境,长时间大电流密度下的工作可能导致活性材料的降解和聚集。为了克服该缺点并达到暴露更多活性位点的目的,通常采用可控合成特殊形貌纳米尺寸磷化钴复合材料的策略,典型代表为碳基材料复合的磷化钴纳米梭、纳米线、中空纳米球、核壳结构等。元素掺杂对磷化钴材料的催化性能具有明显的提升作用,目前大多与碳基质复合,碳基质通常形成三维立体结构,具有丰富的孔道,而拥有大比表面积的磷化钴活性成分均匀分散在碳基质中,获得了增强的电导率并且得到保护。
超级电容器对材料的导电性和比表面积有较高要求,一般用于超级电容器的磷化钴是通过液相手段合成的,可以通过调节表面活性剂的种类和比例,对材料的形貌进行设计。碳布、泡沫镍等导电物质通常被用作构建多维纳米磷化钴的基底,一可以提高导电性,二不需要添加会影响电容和倍率性能的黏合剂。
对于锂离子电池和钠离子电池,目前的重点在于构建合理结构的磷化钴纳米材料及其复合物,常用的手段是设计特定形貌的纳米级磷化钴,将其与导电材料复合,磷化钴被包覆或镶嵌在多孔坚固的3D 导电材料基质中,以期达到提高电子和离子传输速率、缓解体积变化并防止活性物质聚集脱落的目的。自支撑的磷化钴电极材料也受到了关注,在电池组装过程中常需要加入黏合剂,然而黏合剂对活性材料本身的性能有一定的屏蔽作用,因此在不锈钢、碳布、泡沫镍等导电基质上生长的不需要黏合剂的磷化钴材料也值得进一步关注。
虽然磷化钴材料已经表现出良好的电化学性能,但在纳米级磷化钴的形貌设计和多维立体复合物的结构优化上还有很多工作要做,电催化性能和机理需要进一步深入探究,新一代的磷化钴材料应该着重于更长的寿命和循环稳定性,并且考量工艺简化和成本压缩,以期实现大规模商业化应用。
猜你喜欢
材料保护(2022年10期)2022-12-07
电镀与精饰(2022年10期)2022-10-14
空气动力学学报(2022年1期)2022-03-16
天津医科大学学报(2021年1期)2021-12-05
粉末冶金技术(2021年3期)2021-07-28
空气动力学学报(2021年2期)2021-05-04
粉末冶金技术(2021年1期)2021-03-29
科学大观园(2020年22期)2020-11-30
表面工程与再制造(2019年6期)2019-08-24
World Journal of Diabetes(2019年7期)2019-07-23
