
多灶性运动神经病的临床表现及肌电图特征
2021-04-20 03:00王静雯赵红东时建铨崔小丽
癫痫与神经电生理学杂志 2021年2期
王静雯,赵红东,时建铨,崔小丽
多灶性运动神经病(multifocal motor neuropathy,MMN)是一种少见的、仅累及运动神经的慢性多发性单神经病变,病因尚不清楚,可能与郎飞结结区及结旁区病变相关[1-2]。该病早期以上肢受累多见,临床表现为缓慢进展、不对称、以肢体远端为主的肌无力和肌萎缩,感觉神经多不受累;非嵌压部位多灶性运动传导阻滞(conduction block,CB)而感觉神经传导正常是其特征性电生理表现。40%以上的患者血清抗GM1抗体滴度升高,静脉注射免疫球蛋白(intravenous immunoglobulin,IVIg)为首选治疗[3-5]。近几年虽然人们对该疾病的认识已有所提高,但在国内报道较少。现就我院7例MMN患者的临床、神经电生理特点进行回顾性分析,以提高临床医生对该病的认识和诊断水平。
1 资料与方法
1.1 一般资料
本文选择2016年6月至2019年12月南京医科大学附属南京医院(南京市第一医院)收治的7例MMN患者作为研究对象,其中男4例,女3例。年龄40~74岁,平均50岁;平均病程17个月。入组患者均符合2003年美国电诊断协会制定的MMN诊断标准[6]和2010年欧洲神经病学联盟/周围神经病学会关于MMN的诊治指南[7]。7例患者均以肢体远端不对称无力为首发表现,6例表现为单侧上肢远端无力,1例表现为足下垂,均为慢性病程渐累及肢体近端、双侧及四肢。7例伴有肌萎缩,远端重于近端,上肢重于下肢。1例患者有肢体疼痛症状,1例患者有一过性主观麻木,1例发生首发症状自行缓解情况。肢体无力符合周围神经支配。查体未见颅神经异常,均无感觉障碍。
1.2 方法
1.2.1 神经电生理检查
包括神经传导检测(nerve conduction studies,NCS)和肌电图(electromyography,EMG)检查。使用丹麦产Keypoint型号肌电图仪(Medoc公司),在室温20~28 ℃条件下进行。神经CB的判断采用欧洲神经病学联盟/周围神经病学会关于CB判定的标准。使用表面电极对每例患者双正中神经、尺神经、桡神经、胫神经、腓总神经的运动神经和感觉神经传导进行检测,测定结果参考北京协和医院肌电图室正常值。应用同心圆针电极对患者的第I背侧骨间肌、拇短展肌、指总伸肌、胫前肌、腓肠肌进行检测,观察所检肌肉在完全放松时是否存在异常的自发电位、轻收缩时运动单位电位(motor unit potential,MUP)时限、波幅、位相和发放频率以及大力收缩时MUP募集类型。
1.2.2 实验室检查
7例患者入院后均行血常规、血生化、肿瘤标志物及风湿免疫相关指标等常规检测;5例患者行腰椎穿刺,检测脑脊液常规、生化及抗神经节苷脂抗体及抗神经束蛋白抗体滴度;7例患者检测血清抗神经节苷脂抗体及抗神经束蛋白抗体滴度。
2 结果
2.1 神经电生理检查结果
NCS:28条神经(40%)出现复合肌肉动作电位(compound muscle action potentials,CMAP)波幅降低;23条神经(33%)出现不同程度的运动传导速度(motor conduction velocity,MCV)减慢;4条正中神经(6%)、4条尺神经(6%)、2条胫神经(3%)F波潜伏期延长,8条正中神经(11%)、5条尺神经(7%)、5条胫神经(7%)F波出波率降低,1条正中神经(1%)、1条尺神经(1%)F波未引出;4条正中神经(6%)、3条尺神经(4%)出现轻度波形离散;所有患者均见2条以上的运动神经节段性CB,以发生在上肢的运动神经节段性 CB 和在其肢体远端的运动神经节段性 CB 为著。其中,10条正中神经(14%)、7条尺神经(10%)、3条桡神经(4%)、3条腓总神经(4%)、5条胫神经(7%)表现为非对称性节段性运动CB,以正中神经(14%)及尺神经(10%)检出率最高,其中,4条正中神经(6%)、3条尺神经(4%)、1条腓总神经(14%)存在2处及以上的CB;感觉传导检测均正常。结果详见表1。

表1 7例MMN患者的NCS结果比较
EMG:检查44块肌肉,受累神经支配肌肉27块,可见安静时部分肌肉出现纤颤、正相电位,轻收缩时MUP波幅升高、时限增宽、多项波或募集减少等神经原性受损的表现。结果详见表2,典型病案举例见图1、2。

图1 患者高某某,41岁,女性,NCS及EMG结果。A:NCS检测示左尺神经腕横纹处刺激CMAP波幅3.6 mV,距腕横纹10 cm处近端刺激CMAP波幅1.31 mV,呈传导阻滞;B:EMG检测示左第I背侧骨间肌轻收缩时可见部分MUP偏宽大。

表2 7例MMN患者EMG结果比较
2.2 实验室检查结果
7例MMN患者血常规、血生化、肿瘤标志物及风湿免疫相关指标等常规检测均正常。5例患者脑脊液压力、白细胞数、糖、氯化物水平均在正常范围,仅1例患者脑脊液蛋白水平轻度升高(为400 mg/L),5例患者脑脊液抗神经节苷脂抗体及抗神经束蛋白抗体均阴性。5例患者血清抗GM1 IgM抗体阳性,1例患者血清抗NF186 IgG抗体阳性,1例患者均阴性。结果详见表3。

图2 患者吴某某,48岁,女性,NCS及EMG结果。A:NCS检测示右正中神经肘部刺激CMAP波幅3.8 mV,腋部刺激CMAP波幅1.65 mV,呈传导阻滞;B:EMG检测示右拇短展肌轻收缩时见个别MUP偏宽大。
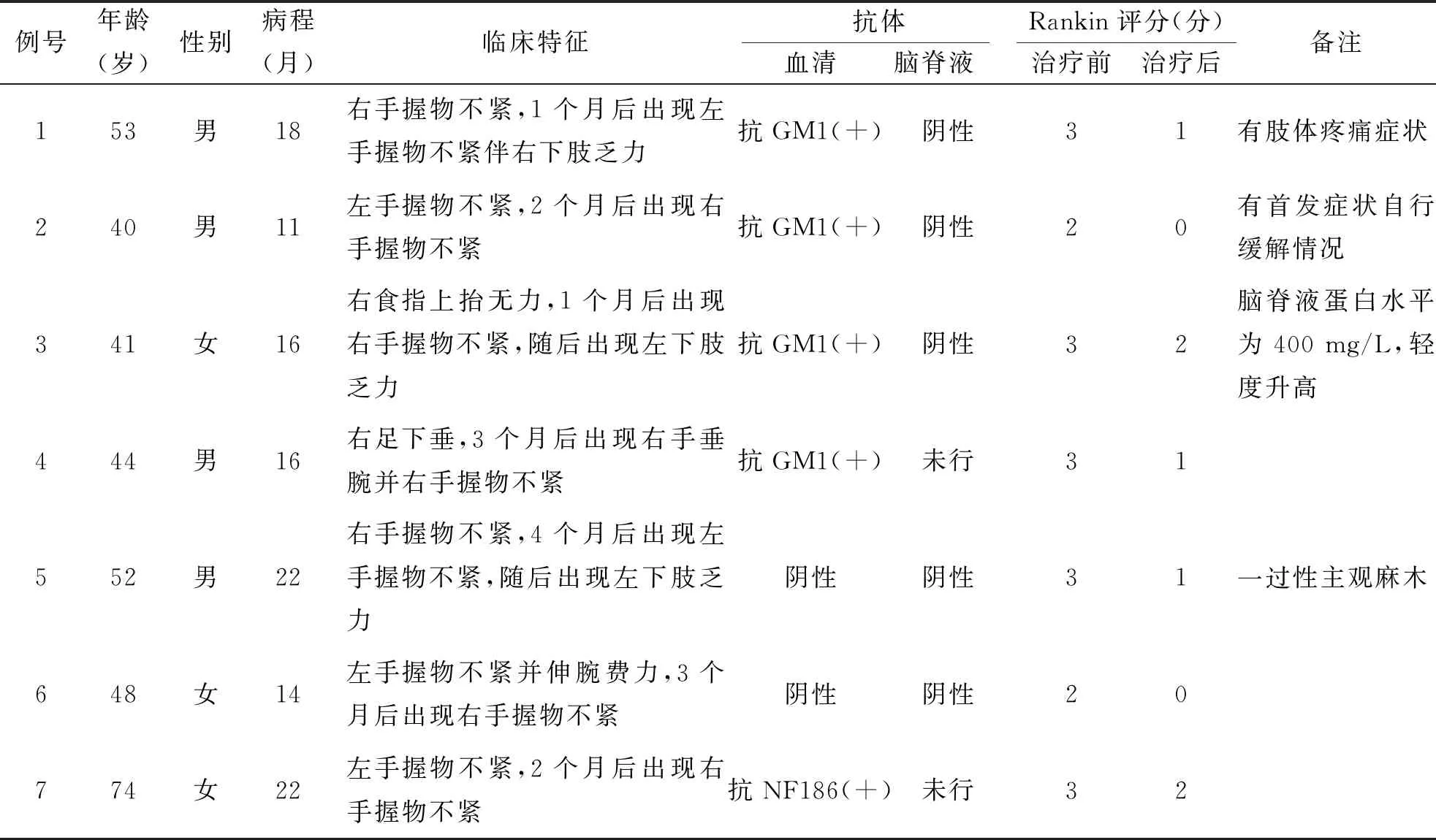
表3 7例MMN患者的临床资料比较
2.3 治疗与预后
入院后均予静脉注射用丙种球蛋白(IVIg)治疗[0.4g/(kg·d)*5 d]。治疗前后均行改良版Rankin残疾评分(表3)。所有患者经治疗1~2周后症状改善。
3 讨论
多灶性运动神经病(MMN)是一种免疫介导的慢性、获得性、局灶脱髓鞘性周围运动神经病,发病率为1~2/100 000,男女比例为2.7∶1,发病年龄为20~70岁,80%的患者50岁前起病[3-4]。大多数患者的症状始于上肢远端,经常出现手握力减弱、手腕下垂或伸指困难,很少出现在上肢近端;约1/3的患者的症状最初出现在下肢远端。本组患者平均年龄50岁,年龄最大者74岁,6例从单侧上肢远端起病,1例从右下肢远端起病,均表现为病情缓慢进展、每例均有两个独立运动神经分布的肌无力,逐渐累及同侧及对侧肢体,并向肢体近端发展,伴肌萎缩,远端重于近端,上肢重于下肢;无感觉障碍,其临床表现与文献报道相似[4,8]。文献报道部分患者可伴有感觉障碍[2,9],本组仅有1例患者有一过性感觉异常主诉,可能与运动神经受累较重而波及感觉神经有关,但因感觉受损较轻,尚未出现EMG改变。
MMN发病机制不明,目前研究认为该病可能与郎飞结结区及结旁区功能异常有关[1-2,4,10]。40%~60%的患者抗GM1抗体阳性[3-4],GM1是神经髓鞘中糖脂的主要组成部分,外周运动神经髓鞘中的含量高于感觉神经,因此MMN以运动神经受累为主。抗GM1抗体与之结合激发的免疫反应导致运动神经局灶性、重度脱髓鞘;此外,抗GM1抗体也可结合在轴索表面,阻止髓鞘再生,导致运动神经出现CB。有研究报道[11]10%的MMN患者抗NF186 IgG抗体阳性,其与郎飞结上的NF186蛋白结合,通过经典的补体途径使得轴突损伤,导致运动神经传导障碍而致病。本组5例患者血清抗GM1抗体阳性,1例患者抗NF186抗体阳性,提示抗体检测对MMN诊断有一定意义;1例患者抗体阴性,可能与抗体滴度较低无法检测到或者存在其他潜在的病理生理机制有关。
本组7例MMN患者EMG均符合周围运动神经病,表现为不对称的运动传导CMAP波幅降低、MCV减慢、F波潜伏期延长或出波率下降,部分患者还可以观察到波形离散,但感觉神经传导均未见异常。每例患者均见有肯定的2条或2条以上的运动神经非嵌压部位至少一处CB,所测神经中以正中神经及尺神经CB检出率最高,分别为14%和10%,发生部位以前臂段最为多见;亦可见单根神经多处CB。非嵌压部位多灶性运动CB而感觉神经传导未受累,是MMN特征性电生理标志[3,9,12]。也有研究报道过没有CB的MMN[13],这可能因CB发生在神经极近端或极远端,受检查技术的局限而未能检测到。CB对MMN不具有特异性,因为弥漫性脱髓鞘导致的“相位抵消”或运动神经慢性轴索病变也可造成CMAP波幅降低,所以在临床EMG操作检测中需要通过“寸移技术”将其与真实的CB区分开来[3]。此外,7例患者存在CB的运动神经支配的肌肉可见不同程度的自发电活动和(或)慢性神经再支配电位,提示MMN是一种以脱髓鞘为主伴轴索变性的周围神经病[14]。
MMN在临床中需与慢性炎性脱髓鞘性多发性神经根神经病(chronic inflammatory demyelinating polyradiculoneuropathy,CIDP)、运动神经元病(motor neuron disease,MND)、遗传性压力易感性周围神经病(hereditary neuropathy with liability to pressure palsy,HNPP)鉴别。CIDP是一种常见的慢性获得性脱髓鞘性周围神经病,往往上下肢均对称受累,并且大多存在感觉传导异常,脑脊液常有蛋白-细胞分离现象,EMG表现为弥散性脱髓鞘改变[14-15]。多灶性获得性感觉、运动周围神经病(lewis-Summer syndrome,LSS)是CIDP一种少见的亚型,因为病灶不对称可能看起来像MMN,但通过患者的临床特点及EMG可资鉴别。与其相比,MMN具有更大的不对称性,不累及感觉神经,进展缓慢,腱反射可正常,脑脊液蛋白多正常或轻度升高。至于MND[16-17]病变则发生在不同脊髓节段的前角而不是单个神经,因而MND患者肌肉萎缩临床表现不符合周围神经支配。此外MND通常累及球部及呼吸系统,具有上运动神经元体征。MND的EMG虽表现为宽大的MUP与自发电位并存的神经原性损害,但初期运动传导往往正常。HNPP[18]是常染色体显性遗传性周围神经病,电生理检查提示易受嵌压部位的传导速度的局灶性减慢,与MMN非嵌压部位的电生理表现不同。
IVIg作为MMN的一线用药[0.4 g/(kg·d)*5 d],可以改善患者肌力及肢体功能,减少CB和轴索变性,但只有20%的患者能够达到长期缓解,大多数患者需要定期使用IVIg[4-5,19-20]。同时,皮下免疫球蛋白( subcutaneous immunoglobulin,SCIg)被认为与IVIg有同样的疗效[5]。有文献报道环磷酰胺对MMN治疗有肯定疗效[4,10]。我们观察到本组患者经IVIg治疗后1~2周内肌无力症状改善。
综上所述,在临床工作中,对于以不对称肢体无力、无感觉障碍为主要症状的患者应考虑到MMN 的可能。电生理检查及抗GM1抗体检测对MMN的诊断有重要意义。IVIg对本病治疗有效。所以早期诊断、治疗对改善预后,提高患者生活品质有积极意义。
猜你喜欢
广西医科大学学报(2021年11期)2021-12-20
糖尿病新世界(2020年16期)2020-11-02
医学新知(2019年4期)2020-01-02
中国社区医师(2019年12期)2019-08-26
中国生物医学工程学报(2019年6期)2019-07-16
基层中医药(2018年11期)2019-01-31
大众健康(2017年1期)2017-04-13
科普童话·神秘大侦探(2017年4期)2017-04-06
中国医科大学学报(2016年11期)2016-12-01
中国药物经济学(2016年2期)2016-03-28
