
乳酸菌基因组代谢网络模型的研究进展
2021-06-07 02:01艾连中侯成杰
食品科学技术学报 2021年3期
艾连中,侯成杰
(上海理工大学 医疗器械与食品学院/上海食品微生物工程技术研究中心, 上海 200093)
随着基因组测序技术的快速发展,基因组测序数据量成指数级增长,大量微生物的基因组序列被公布,人们可以从基因和分子水平对微生物的代谢机理进行深入研究,极大地推动了相关学科的发展。如何有效利用这些高通量数据,获得对生命行为机制的系统理解则成为“后基因组时代”的巨大挑战[1]。生物系统的动态行为是不同性质的多个分子之间非线性相互作用的结果,而计算机模拟可以高效完成这一工作。基于模拟的研究有助于理解生命系统复杂的底层结构,帮助研究人员对生命行为进行预测。而基因组代谢网络模型(genome-scale metabolic models,简称GEMs或GSMM)正是基于这一需求而发展起来的。基因组代谢网络模型是基于特定生物体的基因组序列,将所有与代谢相关的基因、酶、生化反应和代谢物数据整合到一起的知识库,并将其转化为数学模型,实现对目标生物体代谢表型的预测,系统表征基因与表型之间的关系[2]。
基因组代谢网络模型有效地模拟了基因组信息和代谢表型之间的关系,为代谢有关的实验数据提供了坚实的解释框架,并使全细胞代谢的模拟实验变得简单。自1999年世界上第一个基因组代谢网络模型流感嗜血杆菌模型被构建以来[3],截至2019年已有超过6 000个GEMs被构建[4],并被广泛应用于系统生物学[5]、代谢工程[6]、药物开发[7-8]、酶功能预测[9]及微生物群落的相互作用[10]等多个领域。乳酸菌作为一类重要的工业菌,广泛应用于发酵食品工业,特别是乳制品工业,部分乳酸菌因具有益生功能还被应用于医学健康领域。为了更深入地对乳酸菌开展功能机制及代谢相关研究,国内外研究人员已构建了多个乳酸菌的GEMs。这些乳酸菌GEMs的构建使人们可以探索乳酸菌在不同环境中的代谢规律和代谢机理,为乳酸菌的研究和应用提供了有价值的工具。
本文将系统回顾微生物基因组代谢网络模型的构建方法及发展,重点聚焦乳酸菌基因组代谢网络模型的研究进展,并对未来研究趋势进行了展望。
1 基因组代谢网络模型的构建方法
随着基因测序技术和算法的发展,研究人员能够基于特定微生物的基因组测序数据和注释结果构建GEMs。GEMs构建的核心是建立基因- 酶- 代谢反应(GPR)之间的关系。为了完成模型的构建,研究人员需要回答以下问题:酶的底物和产物是什么?是否有多个基因参与同一个酶的表达?每个代谢物的化学计量系数是多少?反应是否可逆?反应在细胞内的定位(细胞质或细胞周质)[11]?这些信息可以通过各种生化数据库、文献以及实验数据来确定。通过建立生化反应组成的网络,就构成了一个针对特定生物体的代谢网络模型。构建过程通常包含4个阶段,见图1。
1.1 构建模型草图
基于基因组注释构建模型草图。构建基因组代谢网络模型的起点是目标生物的基因组注释信息,这些数据可以通过测序得到,也可以在NCBI等公共数据库下载。基因组注释为构建提供了唯一的标识,列出了被认为可能存在于目标生物中的酶,并指出这些基因产物如何相互作用(亚基、复合酶、同工酶)形成具有催化代谢反应的活性酶[11]。代谢数据库(如KEGG[12]、BRENDA[13]、SEED[14]、Transport DB[15])收集了一系列不同生物体中的代谢反应和转运反应,为建立酶与代谢反应的关系提供了重要的参考。基于基因组注释和代谢反应整理所建立的GPR关系就构成了代谢模型的草图。在构建模型的过程中还需要注意,即使酶的EC号相同,不同生物体之间也会存在底物特异性和酶活性不同,因此,酶在目标生物中催化的反应可能不同于数据库中的反应。
1.2 人工精炼
代谢模型草图的人工精炼。代谢模型草图虽然提供了基于基因注释的候选反应合集,但还不能建立目标生物体特有的特征。这些特征信息需要根据目标生物体的文献信息进行人工校正。同时,模型草图中还包含了一些错误反应和代谢缺口(gap),因此必须通过人工精炼以修正错误反应,填补缺失反应。模型中所有反应的化学计量学平衡、电荷平衡、元素平衡都需要进行人工检验。因此人工精炼是整个GEMs构建过程中最耗时耗力的,甚至是烦琐的步骤。代谢网络模型的精炼通常需要数月到一年的时间才能完成,这既取决于目标生物的基因组大小,也取决于目标生物是否有足够数量的生化数据[16]。文献、教材、目标生物特有的数据库以及熟悉目标生物的专家都是人工精炼步骤的重要信息来源。
一个高质量的GEMs是通过基因组注释和人工管理相结合的方式构建的。这一过程将创建一个针对目标生物的生化、基因组、遗传学和表型的知识库。而随着新的基因注释结果和新的实验数据的发表,目标生物的代谢网络模型应以迭代的方式进行更新[17]。
1.3 模型转换
将代谢网络模型转换为数学模型。精炼后的代谢模型需要转换为计算机可以识别的数学格式(S矩阵,如图2),以便通过计算机对模型进行模拟计算。在S矩阵中,行代表网络代谢产物,列代表代谢反应。反应中的底物被定义为具有负系数,而产物具有正系数[16]。
在进行模型模拟计算前,需要定义模型的系统边界,也就是对所有能被目标生物消耗或分泌的代谢物,都需要在模型中加入交换反应。交换反应可以在模拟中用于定义环境条件(如碳源、氧气等)。拟稳态假设和基于约束的重构分析(constraint-based reconstruction and analysis,COBRA)[18]是最广泛应用的模拟计算策略。对模型进行优化计算最常用的目标函数是优化生长速率,即生物量函数,由生长所需的基本代谢产物组成。利用数学表达式和计算平台生成生物质目标函数是验证代谢网络模型最基本的功能。细胞生物质组成数据可以通过实验获得,也可以通过文献或数据库获得。
流量平衡分析(flux balance analysis,FBA)是表征代谢网络模型最常用的方法。FBA是一种分析代谢网络中代谢物流动的数学方法,通过计算代谢网络的代谢物流量,从而可以预测生物体的生长速度或重要代谢物的生成速率[19]。S矩阵和目标函数定义了一个线性方程组,在给定的约束条件下可以求解得到一个解空间。FBA可以识别解空间中优化目标函数的单个最优通量分布或多个最优通量分布。目前,广泛用于流量平衡分析的软件是基于Matlab平台的COBRA工具箱[20]和基于python平台的COBRApy[21]。
1.4 验证评估
模型的验证与评估。精炼后的模型是否能够准确预测微生物的生长表型是需要进行验证和评估的。验证和评估模型的方法有很多,最常用的方法是单一碳源验证、氨基酸缺失验证以及基于文献的各种生理代谢参数验证[22]。例如:通过限制碳源、氨基酸、生长因子的通量,我们可以预测菌株的营养缺陷型;通过恒化培养实验与模拟生长速率进行拟合,可以推测出模型中存在的影响生物质合成的代谢途径[11,16]。将这些模拟计算的结果与“湿”实验数据进行对比验证,可以帮助我们分析模型中的缺陷并改进。总之,代谢网络模型的构建是一个不断迭代的过程,而如何确定模型构建完成则取决于模型构建的范围和用途[16]。
为了确保构建结果的准确性和可用性,Palsson课题组于2010年发表了构建代谢网络模型的标准化操作程序,该程序共分为5个阶段,包含了96个具体步骤[16]。该程序很好地规范了构建GEMs的流程及GEMs的格式,使构建GEMs具有了统一的标准,方便国际间的交流与合作。
人工构建基因组代谢网络模型费时费力,因此多个研究团队还开发了用于GEMs构建的自动化工具,如ModelSEED[23]、RAVEN[24]、Merlin[25]、CarveMe[26]、kbase[27]等。这些自动化工具大大提高了构建GEMs 的效率,有的工具只需要几十个小时就可以构建一个GEMs。利用自动化工具构建的GEMs普遍存在大量的错误反应、代谢缺口,甚至是基因注释错误,所以这些自动构建的GEMs通常只能作为代谢模型的草图使用,还需要人工精炼才能成为高质量的GEMs。现有的自动化构建工具尚无法完全替代人工精炼的步骤,目前最常用的方法是自动化构建与人工精炼相结合的方式,即先通过自动化工具构建出目标生物的代谢网络模型草图,再由人工对草图进行精炼和验证,最终得到一个高质量的GEMs[28-29]。
2 微生物基因组代谢网络模型的发展历程
基因组测序数据的指数级增长加速了微生物GEMs的构建,截至2019年2月,已有6 105个微生物基因组代谢网络模型被构建,其中细菌5 897个,古细菌127个,真核微生物81个[4]。这些快速增加的GEMs中大部分是由自动化工具构建的未人工精炼模型,虽然也被用于各种微生物的研究,但相较于经过人工精炼的高质量GEMs,它们的预测准确度比较差。以大肠杆菌为代表的模式生物,因被科学家广泛研究而积累了大量生理生化数据,使得这些模式生物在构建GEMs的过程中具有极大的优势,产生了一系列高质量的GEMs。以大肠杆菌为例,从2000年发布的第一个模型开始到2017年发表的iML1515[30],已经历了至少6次迭代,模型的规模和准确度都有了极大提升。这些模式菌株GEMs的构建和发展使代谢网络模型的应用不断扩大,为目标生物体的代谢研究提供了极好的知识库,同时也为其它微生物GEMs的构建提供了良好的参考模板,推动更多模型的出现,也推动了构建方法的不断升级。
2.1 大肠杆菌基因组代谢网络模型
第一个大肠杆菌(Escherichiacoli)基因组代谢网络模型是基于大肠杆菌E.coliK-12 MG1655构建的iJE660[31],该模型包含了660个基因,含有完整的氨基酸、核酸、细胞壁和辅因子合成代谢、脂肪酸代谢,能够对生长表型进行预测,基因缺失模拟的准确度为86%。在后续的几次迭代中,陆续添加了不同碳源代谢、醌的表征[32]、细胞周质交换反应、完善了元素平衡和电荷平衡,并填充了缺失反应[33],不断提高模型的准确性,见图3。最新的iML1515模型包含了1 515个基因,2 719个代谢反应和1 192个代谢物,并加入了所有酶的三维结构。iML1515对基因重要性的模拟准确率达到了93.4%。此外,iML1515还可以从大量生物数据中提取最相关的信息快速生成新的子模型。例如:通过iML1515生成的子集iML976仅包含了1 000多个大肠杆菌菌株共有的代谢网络信息,使人们可以更清楚的了解大肠杆菌的核心和辅助代谢能力,突出了识别药物靶标的潜力。将iML1515用于分析不同条件下的转录组数据为转录组变异分析提供了有价值的见解。由大肠杆菌GEMs的发展可以看出,随着模型的不断迭代,其覆盖的基因和代谢反应数量不断增加,模型的应用范围也不断扩大。早期的GEMs主要用于计算微生物的生长速率、副产物产量等基本表型,而最新的GEMs已经可以用于各种应激反应分析、泛基因组分析、蛋白质组功能分析等[30]。
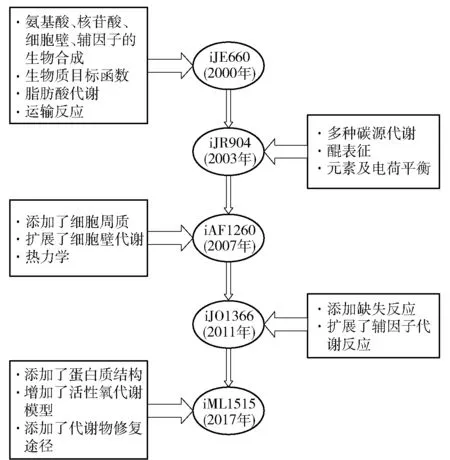
图3 大肠杆菌GEMs的迭代Fig.3 Iteration of E. coli GEMs
2.2 枯草芽孢杆菌基因组代谢网络模型
枯草芽孢杆菌(Bacillussubtilis)是革兰阳性菌中的代表性菌种,具有出色的生产异种蛋白的能力,被认为是工业酶和生物制药的“细胞工厂”[34]。目前已有多个枯草芽孢杆菌的GEMs被构建,包括iYO844[35]、iBsu1103[36]、iBsu1103V2[37]、iBsu1147[38]、iBsu1144[39]以及天津大学研究团队构建的iBsu1141[40]。最新版本的枯草芽孢杆菌GEMs是iBsu1144,该模型基于对枯草芽孢杆菌基因组重新注释而建立。iBsu1144包含了1 144个基因、1 955个反应和1 103个代谢物,通过热力学分析重新修正了代谢反应的方向性和可逆性。基于必需基因和非必需基因分析的准确率分别达到91.6%和91.4%。该模型可用于枯草芽孢杆菌的代谢工程设计,同时也为其他革兰阳性菌GEMs的构建提供了参考模板。
2.3 酿酒酵母基因组代谢网络模型
酿酒酵母(Saccharomycescerevisiae)是第一个被构建GEMs的真核微生物[41]。与原核微生物不同,真核微生物中存在多个细胞器,在构建GEMs时需要考虑代谢反应发生的区室并加入胞内转运反应,因此真核微生物GEMs的构建过程更加复杂。
酿酒酵母作为真核生物的模式生物而受到广泛的关注和研究,从酿酒酵母的第一个GEMs[41]诞生以来,已经有多个研究团队发布了多个版本的酿酒酵母GEMs[42-44]。由于不同研究团队在注释结果和术语等方面存在差异,使不同模型之间的比较和使用都变得困难,给酿酒酵母GEMs的应用和升级带来了极大困扰。为解决这一问题,科学界通过国际合作建立了一个关于酵母的共识代谢网络模型[45],这一共识模型采用统一的规范术语并由专人维护升级。最新发表的酿酒酵母代谢模型有效整合了RNA和蛋白质合成数据,得到了第一个添加了表达与热力学通量(ETFL)的酿酒酵母代谢与表达模型(ME-models)。在预测最大生长速率、必需基因和溢出代谢表型等方面表现出良好的能力[46]。
3 乳酸菌基因组代谢网络模型的构建现状
乳酸菌是一类能够将碳水化合物转化为乳酸的细菌的统称[47]。乳酸菌在发酵的过程中利用碳源产生乳酸使食品快速酸化,可以延长食品的货架期和保质期,同时乳酸菌代谢产生的一些化合物还能够赋予食品特殊的风味和质地[48]。近二十年来,随着微生物组技术、组学技术和生物信息学技术在乳酸菌领域的应用,人们对乳酸菌的代谢有了深入的认知,构建乳酸菌的GEMs也有助于更深入的研究乳酸菌的代谢活动,指导乳酸菌的代谢工程改造。表1列出了已发表的乳酸菌GEMs[28-29,49-55]。
3.1 乳酸乳球菌基因组代谢网络模型
乳酸乳球菌(Lactococcuslactis)作为乳酸菌的模式菌株,是研究最广泛的一种乳酸菌,其基因组也是乳酸菌中第一个被测序和注释的[56]。第一个乳酸乳球菌GEMs是由Nielsen课题组于2005年构建[49]。该模型是基于Lactococcuslactisssp.lactisIL1403的基因组注释构建的,包含358个基因、621个反应和509个代谢物,其中476个代谢反应与基因建立关联,其余145个代谢反应是基于生理生化考虑而推测加入的。通过FBA和MOMA进行代谢分析,证明该模型的很多预测结果和实验结果是吻合的。例如:在所有氨基酸都被提供的情况下,乳酸乳球菌更倾向于从头合成丙氨酸、天冬氨酸、甘氨酸和苯丙氨酸。该模型作为一个有用的工具可用于测试或开发新的代谢工程策略[49]。
另一个乳酸乳球菌GEMs是由荷兰瓦赫宁根大学的研究团队基于Lactococcuslactisssp.cremorisMG1363构建的,得益于十几年来分子生物学和基因组学的快速发展,该模型涵盖的基因数量明显增加,包括518个基因、754个反应和650个代谢物。通过FBA和FVA分析了整个代谢网络中的通量分布情况以及与风味物质形成有关的通路,发现754个代谢反应中有59个反应直接或间接参与了风味的形成。全面的模型驱动分析显示了乳酸乳球菌高度灵活的氮代谢,与氧化还原平衡相结合的支链氨基酸分解代谢是预测不同风味化合物形成的关键。该模型为解析乳酸菌合成风味物质的代谢网络提供了基础工具[50]。
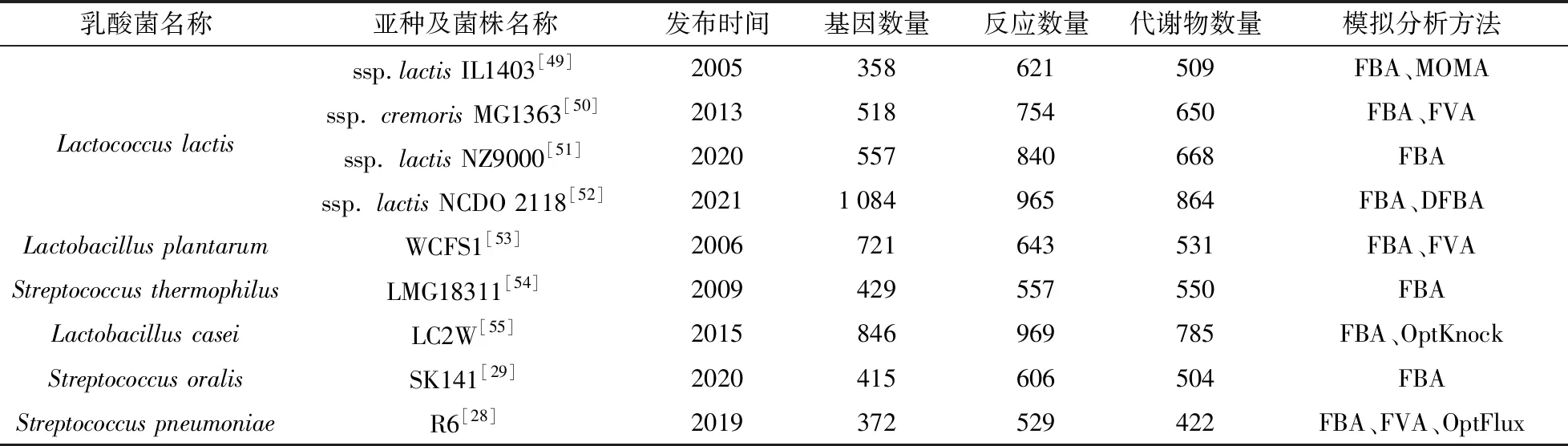
表1 已发表的乳酸菌GEMsTab.1 Published lactic acid bacteria GEMs
最新发表的乳酸乳球菌GEMs(iOA1084)是基于Lactococcuslactisssp.lactisNCDO 2118构建的,该菌株具有高产γ-氨基丁酸(GABA)的能力。iOA1084涵盖了62个代谢途径、1 084个基因、965个代谢反应和864个代谢物。研究者重点关注了通过模型预测提高菌株产GABA的能力,通过FBA分析正常和高谷氨酸摄取速率下GABA的产生,发现高谷氨酸可以增加GABA的生产速率[52]。
3.2 植物乳杆菌基因组代谢网络模型
植物乳杆菌(Lactobacillusplantarum)是一种工业菌株,因其具有益生作用而受到广泛关注。研究人员基于植物乳杆菌WCFS1构建了一个包含721个基因、643个反应和531个代谢物的GSMs,优化了ATP的产生并确定了与自由能代谢无关的氨基酸分解代谢途径。通过FVA分析补充了基本模型中的平行途径,如产物相同但辅因子不同的途径。FBA分析显示模型过高估计了增加葡萄糖浓度产生的生物质通量而低估了乳酸生成量,这可能是因为模型假定细胞生成生物质的效率最优,所以不能有效预测代谢效率较低的乳酸的生成[53]。
3.3 嗜热链球菌基因组代谢网络模型
嗜热链球菌(Streptococcusthermophilus)作为优良发酵剂被广泛用于乳制品工业,基于嗜热链球菌LMG18311的GEMs于2009年发布并与其他两种乳酸菌(乳酸乳球菌和植物乳杆菌)的GEMs进行了比较。相较于其它两种乳酸菌,嗜热链球菌显示出较低的自养能力,这种低自养能力是由于嗜热链球菌的进化环境决定的。此外,风味分析发现嗜热链球菌氨基酸代谢通路产生了较多的挥发性化合物,这也是其赋予酸奶等发酵乳制品特殊风味的原因[54]。
3.4 干酪乳杆菌基因组代谢网络模型
干酪乳杆菌(Lactobacilluscasei)被认为是一种具有益生作用的乳酸菌,在乳制品、制药及临床医学领域均有应用。江南大学刘立明团队利用人工注释和ModelSeed自动建模结合的方式构建了第一个干酪乳杆菌基因组代谢网络模型iJL846,该模型由846个基因、969个代谢反应和785个代谢产物组成。模型分析发现有10种氨基酸和7种维生素是干酪乳杆菌LC2W的必需营养素;通量分析表明EMP途径是产生乳酸的主要途径,同时预测了混合酸发酵过程中氨基酸的通量。FBA分析表明挥发性风味化合物的形成与氧环境有直接关系,并通过模拟基因缺失预测了三个可以改善3-羟基丁酮产量的新靶点[55]。
3.5 口腔链球菌基因组代谢网络模型
口腔链球菌(Streptococcusoralis)是一种存在于口腔中的乳酸菌,与口腔中其他细菌存在着复杂的互作关系,这些互作关系被认为与人类的口腔健康有关。Palsson课题组基于口腔链球菌SK141的基因组序列,并结合近缘物种的实验数据构建了首个口腔链球菌的GEMs(iCJ415)。利用近缘物种的基因必需性数据和氨基酸营养缺陷数据验证该模型,其基因必需性的预测准确率为71%~76%,氨基酸营养缺陷预测准确率为85%,碳源预测结果的准确率为82%。说明iCJ415可以较好地反映口腔链球菌的代谢特性,该模型可以作为探索同一物种不同菌株之间相互作用以及不同物种在人类口腔内复杂代谢作用的起点[29]。
3.6 肺炎链球菌基因组代谢网络模型
肺炎链球菌(Streptococcuspneumoniae)是一种革兰氏阳性菌,可以产生少量乳酸,因此也属于乳酸菌[47]。但不同于大多数乳酸菌,肺炎链球菌是一种致病菌,可导致肺炎、中耳炎等疾病。为了更好地了解肺炎链球菌的代谢,研究人员利用自动化构建软件Merlin构建了肺炎链球菌的基因组代谢模型iDS372,该模型可以模拟肺炎链球菌在不同环境条件下的代谢行为,为新的药物靶点提供了线索[28]。
4 乳酸菌基因组代谢网络模型的研究展望
虽然研究人员很早就开始了乳酸菌GEMs的构建,但相较于以大肠杆菌和酿酒酵母为代表的模式菌株,乳酸菌GEMs无论在数量还是质量上都明显落后。很多模型是十几年前构建的,模型中往往只包含了GPR关系等核心内容,缺少代谢通路子系统分类,代谢物也没有进行统一术语规范,不利于其他研究人员对模型进行应用和升级。乳酸菌GEMs的研究还有很大的空间,同时也存在巨大的挑战。未来乳酸菌GEMs的研究可以重点围绕以下两个方面开展。
4.1 更新乳酸菌GEMs,构建乳酸菌ME模型
随着基因组学、微生物组学的快速发展以及大量乳酸菌生理生化数据的更新,基因注释的范围及准确性都有了明显提高,而现有的乳酸菌GEMs构建时间普遍较早,无论是基因注释信息、数量以及代谢反应的数量和质量都需要进行更新。随着蛋白组、转录组、代谢组等组学技术的发展,整合代谢模型和蛋白质合成途径(包括转录、翻译)的ME模型(代谢与大分子表达模型)应运而生。ME模型除了反应代谢网络外,还明确包含了构成转录和翻译的途径,能够模拟蛋白质组的组成,因此,ME模型可以用于计算菌株生长条件或新环境进化适应性的蛋白质组分配,大大扩展了模型的功能和应用范围。ME模型是代谢模型(M)和表达矩阵(E)的整合,表达矩阵中包含了已知的所有功能成分(蛋白质、核苷酸等)和转录途径,包括RNA和蛋白质的合成、修饰和降解[17]。ME模型不仅可以预测细胞的最大生长速率和相应的代谢通量,还可以计算最优的蛋白质组分配和基因产物表达水平。通过ME模型可以实现组学数据的定量集成和表型模拟。目前已经发表了多个大肠杆菌ME模型[9, 30],但是还没有针对乳酸菌的ME模型。构建乳酸菌ME模型可以为乳酸菌的代谢工程设计提供更有价值的策略。
4.2 构建乳酸菌泛基因组模型
随着基因组测序数量的不断增加,泛基因组的概念被提出。泛基因组是指某一物种全部基因的总和,由核心基因组(即一个物种内所有菌株共有的基因)和辅助基因组(即只存在于某个菌株的基因)构成[57]。泛基因组可用于不同菌株之间的基因比较分析,有助于对菌株代谢过程的所有机制进行更深入的分析。通过对炎症性肠病患者临床分离得到的大肠杆菌进行泛基因组分析发现,患者体内分离到的特定大肠杆菌菌株能通过特殊代谢途径参与黏液多糖代谢,有助于该菌株在肠道内的定植[58]。乳酸菌在肠道中的定植能力与其功能作用有着密切的关系,大肠杆菌的泛基因组分析为乳酸菌在肠道内的定植研究提供了新思路。目前已构建的泛基因组代谢网络模型主要集中于大肠杆菌、沙门氏菌[59]、金黄色葡萄球菌[60]等致病菌。而乳酸菌中包含数量众多的益生菌,其在维持人体肠道健康等方面具有重要的作用,建立乳酸菌泛基因组模型将有助于我们解析这些菌株的特异性,研究其在人体肠道定植以及与其他肠道微生物互作的机理。
猜你喜欢
四川生理科学杂志(2022年4期)2022-12-06
军事文摘(2022年16期)2022-08-24
河南畜牧兽医(2021年9期)2021-12-10
今日农业(2021年11期)2021-08-13
皮肤病与性病(2021年3期)2021-07-30
中国现代医药杂志(2020年10期)2020-12-14
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
小学阅读指南·低年级版(2016年10期)2016-09-10
食品与健康(2015年9期)2015-09-08
