
9例色素异常性皮肤淀粉样变患者GPNMB基因序列分析
2021-07-24 09:34冯婉婷吴芳芳陈俊溢杨斌
皮肤性病诊疗学杂志 2021年3期
冯婉婷,吴芳芳,陈俊溢,杨斌
1.广东医科大学第一临床医学院,广东 湛江 524000;2.南方医科大学皮肤病医院,广东 广州 510091
原发性皮肤淀粉样变(primary cutaneous amyloidosis,PCA)是指淀粉样蛋白局限性沉积于真皮浅层,而不累及其他器官的一种慢性瘙痒性皮肤病[1]。临床上最为常见的两型为苔藓性淀粉样变和斑状皮肤淀粉样变[2]。色素异常性皮肤淀粉样变(amyloidosis cutis dyschromica,ACD)是PCA的罕见类型,通常发生在青春期前,好发于躯干和四肢,表现为对称分布的点状或网状的色素沉着,其间可见色素减退斑,伴或不伴有瘙痒;组织病理学表现与PCA大致相同,均为真皮浅层的淀粉样物质沉积[3]。2018年Yang等[3]证实ACD的致病基因是GPNMB[MIM:604368],不同于PCA常见类型的致病基因OSMR和IL-31RA[1]。GPNMB基因编码的非转移性黑色素瘤糖蛋白B(glycoprotein non-metastatic melanoma protein B,GPNMB)几乎表达于所有表皮细胞中,以黑素细胞最为显著[4],其次是角质形成细胞[3]。此后,关于GPNMB基因突变的ACD病例被陆续报道,且遗传模式具有多样性。为验证GPNMB在ACD患者中的致病情况,本研究对9例经皮肤病理确诊为ACD的汉族患者进行GPNMB基因测序,现将结果报道如下。
1 资料与方法
1.1 研究对象
2015—2019年于我院门诊经病理确诊[5]的9例ACD患者。收集患者的临床表现、家族史、病理结果等资料。所有患者及家属均签署知情同意书。
1.2 方法
1.2.1 DNA提取收集9例先证者及部分家属的外周血各2 mL(EDTA抗凝),采用TIANamp Blood DNA试剂盒提取DNA,并用Nanodrop分光光度计进行质检。双链DNA浓度>30 ng/μL,OD260/OD280比值在1.7~1.9范围内,视为样品合格。
1.2.2 GPNMB基因检测GPNMB基因12个外显子的特异引物序列参照既往文献报道[6]。采用30μL PCR反应体系,Bio-Rad PCR仪进行扩增。扩增程序为96℃预变性5 min;96℃变性20 s,62℃退火20 s,72℃延伸30 s;以后每个循环依次降低1℃,直到51℃,共11个循环后96℃变性20 s,52℃退火20 s,72℃延伸30 s,共34个循环;72℃延伸5 min。所得PCR产物经1%琼脂糖凝胶电泳、凝胶成像及条带分析后,确定产物的目的片段,切胶回收送至测序公司进行纯化和测序分析。测序结果与Genbank中人类GPNMB基因的标准序列进行比对后,采用MutationTaster预测突变位点的致病性。
2 结果
2.1 一般资料
9例ACD患者中男5例,女4例,发病年龄2~47岁,中位年龄23岁。其中2例女性患者青春期前发病,2例女性、1例男性青春期发病。
2.2 临床特征
9例患者皮损表现大致相同,均好发于四肢伸侧,表现为四肢和(或)躯干弥漫的色素沉着,其间可见散在大小不一的色素减退斑(图1A、1B)。5例初发皮损为四肢伸侧的散在白色斑点,4例具有水疱的患者初发症状则为四肢伸侧的小水疱。9例均未见毛细血管扩张和皮肤萎缩;甲、毛发、牙齿、黏膜及掌跖均未受累;全身系统检查无明显异常。
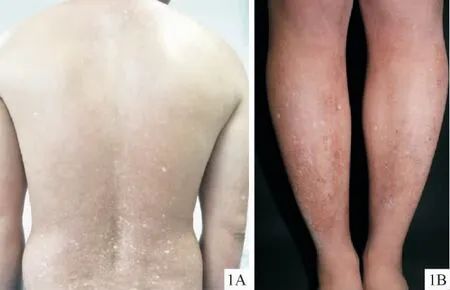
图1 ACD患者Ⅴ躯干(1A)、患者Ⅷ下肢(1B)色素沉着伴色素减退Figure 1 Clinical manifestation of ACD:hyperpigmentation and hypopigmentation of the trunk(1A:patientⅤ)or limbs(1B:patientⅧ).
6例患者轻度瘙痒,其中4例仅在水疱出现前局部有瘙痒症状,且水疱的进展具有一定的规律,即初起为四肢伸侧局部瘙痒;随后于瘙痒处出现红豆大小的水疱,疱液清亮,可为散在或线状分布;数天后水疱干涸,瘙痒减轻自行结痂,痂片脱落后遗留与水疱同等大小的色素减退斑,周围遗留色素沉着斑,可反复发作数年后趋于稳定。3例女性患者诉妊娠和(或)哺乳期水疱数目增多,后可自行缓解。水疱最多时可于双上肢出现30个,双下肢出现20个。6例症状可于日晒后加重,表现为色素沉着斑颜色加深。详见表1。
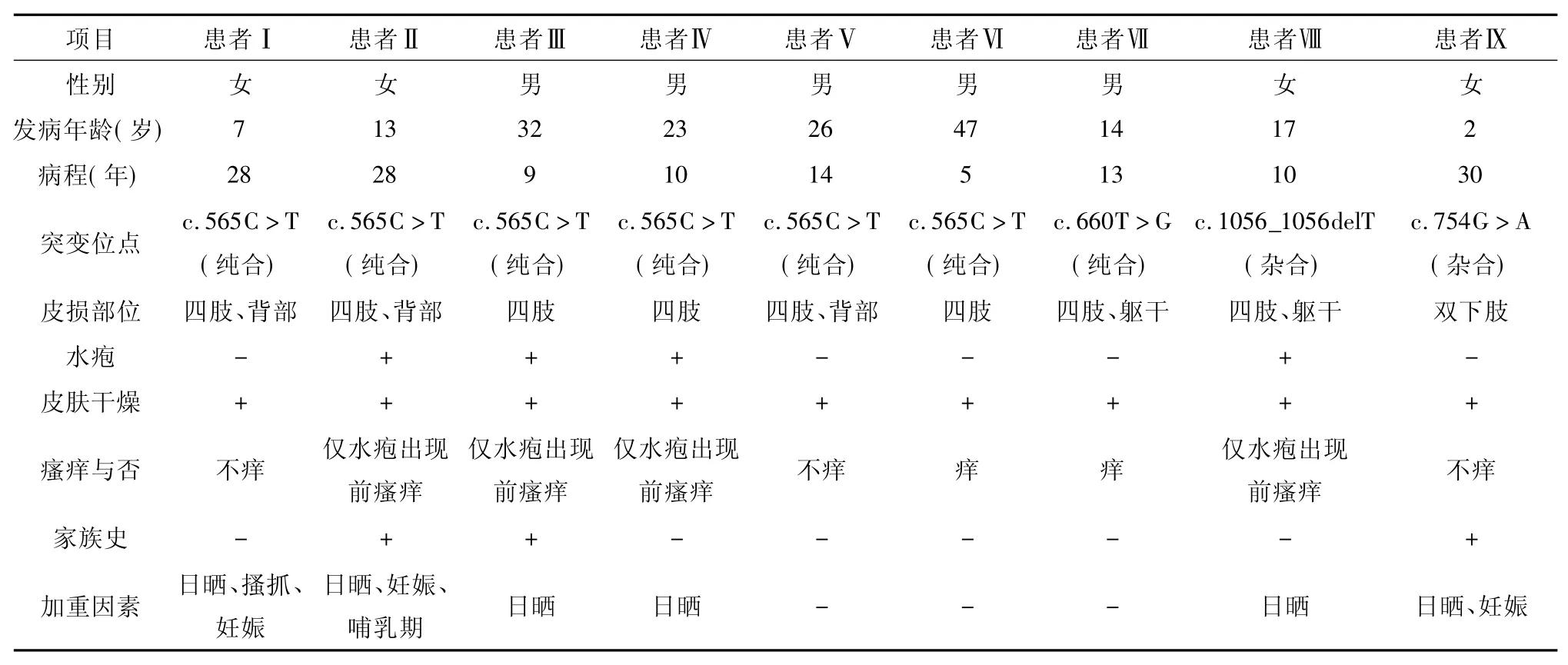
表1 9例ACD患者的临床资料Table 1 Clinical and genetic data of 9 patients with ACD
2.3 家族史
患者Ⅱ和患者Ⅲ为同父同母的兄妹,父母非近亲结婚。患者Ⅸ自诉其外婆、母亲、弟弟均有类似皮肤表现。以上2个家系中的患者均无身材矮小、光过敏、智力下降、掌跖角化以及长期暴晒史。其余患者家族中未见类似病史。
2.4 病理表现
9例均行组织病理检查,HE染色可见表皮突增宽或下延,真皮乳头层见多个灶状的嗜伊红均质物质沉积,血管周围少许淋巴组织细胞浸润(图2A)。刚果红染色可见真皮乳头层橘红色的淀粉样物质沉积(图2B)。
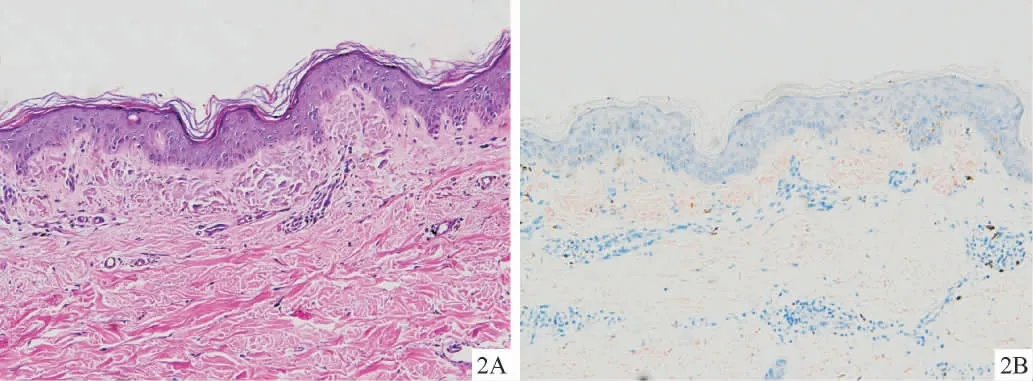
图2 ACD病理表现2A:表皮基底层色素细胞增多,真皮乳头层见多个灶状的嗜伊红均质物质沉积,血管周围少许淋巴组织细胞浸润(HE染色,200×);2B:真皮乳头层橘红色的淀粉样物质沉积(刚果红染色,200×)Figure 2 Histopathology of amyloidosis cutis dyschromic.2A:HE staining shows increased melanocytes in the epidermal basal layer,multiple focal eosinophilic homogeneous deposits in the dermal papilla along with perivascular infiltration of lymphocytes(200×);2B:Congo red staining shows the orange amyloid deposits in the dermal papilla(200×).
2.5 治疗与预后
9例患者治疗均以对症处理为主,予抗组胺药止痒,辅以维A酸乳膏及维生素E乳膏外用以淡化色素。2例在此基础上联合应用果酸治疗,3次后,色素沉着斑较前减轻。
2.6 GPNMB基因测序结果
患者GPNMB基因外显子突变位点见图3。6例患者(Ⅰ-Ⅵ)为c.565C>T纯合突变;其中,患者Ⅰ-Ⅳ的父母测序结果显示均为杂合突变,但查体未见有ACD的典型皮损表现,遗传模式符合常染色体隐性遗传。其余为c.660T>G纯合突变(患者Ⅶ)、c.1056_1056delT杂合突变(患者Ⅷ)、c.754G>A杂合突变(患者Ⅸ)各1例。
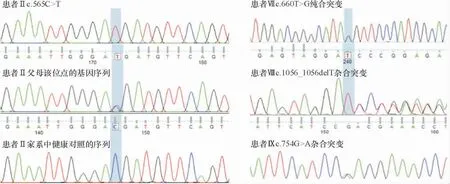
图3 GPNMB基因突变位点Figure 3 The GPNMB gene mutation sites.
3 讨论
ACD在东亚、东南亚种族中较为常见[3,7]。1970年由日本学者Morishima等[8]首次报道并总结特征如下:①全身点状或网状色斑沉着伴色素减退;②轻度或无瘙痒症状;③青春期前发病;④真皮乳头层可见局灶性淀粉样沉积。皮疹进展缓慢,呈对称分布,发病无性别倾向[7]。因其具有异色样的皮损改变,需与着色性干皮病、先天性角化不良、遗传性对称性色素异常症、遗传性泛发性色素异常症、Dowling-Degos病等鉴别[7,9],可依靠病理活检确诊。需要注意的是,皮肤异色病样淀粉样变性(poikiloderma-like primary cutaneous amyloidosis,PCA)与ACD在临床表现、病理特征上极为相似,均为皮肤异色病样损害和淀粉样物质的局限性沉积[10]。区别点在于前者可有苔藓样丘疹,表皮萎缩,毛细血管扩张;并可伴有光敏性、身材矮小、水疱、掌跖角化[11],而后者多为弥漫的色素沉着及减退,无丘疹及掌跖角化[10]。近年来有文献报道ACD也可出现水疱[3],需仔细鉴别。本研究中有4例患者出现过水疱,并具有一定发病规律:瘙痒-水疱-结痂-色素减退。
ACD患者常有家族史,既往认为其可能为外显不全的常染色体显性遗传[7]。随着ACD致病基因的明确,关于ACD患者GPNMB基因突变的病例被陆续报道,丰富了ACD的遗传模式。目前已证实的遗传模式有常染色体隐性遗传[3,12]和半显性遗传[6]。半显性遗传即杂合子的表型介于显性纯合子与隐性纯合子的表型之间,携带杂合子的ACD患者可仅表现为较轻的皮肤色素异常,而无淀粉样蛋白沉积[6]。
目前关于中国汉族患者的报道均符合常染色体隐性遗传,已发现的突变位点有c.565C>T的纯合突变[3,13],或分别与c.660T>G(ex 5)、c.1056del(ex 7)、c.296del(ex 3)共同构成复合杂合突变[3],以及3例复合杂合突变,即c.393T>G和c.717_718del、c.393T>G和c.413G>A[12]、c.719_720del和c.877_880del[3]共同构成的复合杂合突变。复合杂合突变即当患者同时具备两个分别来源于双亲的杂合突变时,可有疾病的临床表现;单独携带其中一个突变位点,则无临床表现[14]。本研究对9例ACD患者进行了GPNMB外显子测序,尚未发现复合杂合突变的现象。有6例均为c.565C>T纯合突变,其中4例经验证符合常染色体隐性遗传。此外,c.660T>G纯合突变、c.1056_1056delT杂合突变、c.754 G>A杂合突变各1例。由于患者Ⅸ(c.754G>A突变)家庭中连续三代有ACD疑似病例,推测该家系可能为常染色体显性遗传。c.754G>A突变位点经评估为本研究中的新发致病位点,但由于家系的失访,无法作进一步家系验证,有待更多报道加以证实。结合既往关于中国汉族患者的报道[3,12-13],GPNMB基因c.565C>T在ACD患者的突变率为10/16,是GPNMB基因的高频突变位点,而该位点在中国人群中的等位基因频率为0.00160559[15]。
综上所述,本研究显示位于GPNMB第5号外显子的c.565C>T可能为我国ACD患者的常见突变位点,且c.565C>T突变位点在遗传模式上具有多样性,与表型的关系具有一定的复杂性;研究发现的GPNMB基因新突变c.754G>A扩展了基因突变谱,但该突变位点的致病性及功能仍有待进一步研究。
猜你喜欢
表面技术(2022年9期)2022-09-27
辐射研究与辐射工艺学报(2022年4期)2022-08-29
临床骨科杂志(2022年3期)2022-06-24
汽车工程师(2021年12期)2022-01-18
爱你·健康读本(2019年8期)2019-11-22
爱你(2019年29期)2019-11-07
环球市场信息导报(2018年21期)2018-07-27
考试周刊(2017年26期)2017-12-12
校园英语·下旬(2017年7期)2017-07-14
科技视界(2016年27期)2017-03-14
