
SPE-HPLC 法快速测定食品中的米酵菌酸及其膳食风险评估
2021-09-14 00:49钟玉心陈悦铭黄景初彭名军苏燕瑜陈嘉欣蔡伟谊
食品工业科技 2021年17期
钟玉心,陈悦铭,王 宇,黄景初,彭名军,苏燕瑜,陈嘉欣,蔡伟谊
(广州市食品检验所,广东广州 511405)
米酵菌酸(Bongkrekic acid, BKA),也叫黄杆菌毒素A,是椰毒假单胞菌属产生的一种线粒体毒素,其化学结构较为稳定(见图1),主要存在于谷类发酵制品、薯类制品、银耳、黑木耳及椰子发酵制品等,难以去除[1−3]。研究表明,米酵菌酸通过抑制线粒体ATP 的合成,损害肝、脑以及肾脏,人食用了被米酵菌酸污染的食物会导致严重的食源性疾病[4−7]。近年来,米酵菌酸中毒事件时有发生,给消费者的身体健康造成重大威胁[8−11]。

图1 米酵菌酸的化学结构Fig.1 Chemical structure of bongkrekic acid
在GB 7096-2014《食品安全国家标准 食用菌及其制品》中,规定银耳及其制品中米酵菌酸含量不能超0.25 mg/kg[12],但未对其他食品作出明确的限量要求。有研究表明,摄入1~1.5 mg 米酵菌酸可使人致命[11]。由于缺乏相应技术规范,在生产过程中容易造成米酵菌酸超标,导致米酵菌酸中毒事件频发[13−14],因此,有必要对市场流通的银耳及其制品、酵米面及其制品和发酵型饮料等食品中米酵菌酸的含量进行检测,并对其膳食暴露风险进行评估。为此,建立一种准确、快速、高效的米酵菌酸检测方法,对加强食品安全监管尤为关键。
目前,米酵菌酸的测定方法主要有荧光免疫层析法[15]、高效液相色谱法[16−20]、液相色谱质谱法[21−24]等。其中荧光免疫层析法方便快检,适用于现场快速筛查,但其假阳性率较高,难以满足监管执法要求;质谱法灵敏度高、检出限低,但设备昂贵,难以推广。我国现行的检测标准《GB 5009.189-2016食品安全国家标准 食品中米酵菌酸的测定》[25]只适用于银耳及其制品、发酵米面及其制品等样品,基质类别覆盖面小,而且液液萃取法需要消耗大量高毒性的有机溶剂,对环境不友好,回收率低[17-18]。
本研究拟建立一种SPE-HPLC 法快速测定食品中米酵菌酸含量的方法,并结合膳食暴露风险评估技术,对不同类别食品中米酵菌酸的含量展开初步风险评估,以期客观、科学地评估食品中米酵菌酸的含量状况,为食品安全生产及政府相关部门的监管提供科学参考。
1 材料与方法
1.1 材料与仪器
米酵菌酸标准品 纯度≥99.9%,上海安谱实验科技股份有限公司;甲醇 LC/MS 级,美国Thermo公司;氨水(优级纯)、甲酸、乙酸、乙腈(色谱纯)、无水硫酸钠、氯化钠、无水硫酸镁(分析纯) 天津科密欧化学试剂公司;米粉、银耳、玉米粉、椰子发酵饮料样品 均购自广州市某农贸市场。
Waters 2695 高效液相色谱仪(配PDA 检测器)、Oasis HLB 固相萃取柱(500 mg/6 mL)、XSelect HSS T3(4.6×250 mm, 5 μm) 美国Waters 公司;CP225D 电子天平 德国赛多利斯公司;TurboVap氮吹仪 美国Caliper 公司;MIlli-Q Academic 超纯水系统 德国默克密理博公司;MA3basic 圆周震荡器 德国IKA 公司;台式高速冷冻离心机3-18 KS德国SIGMA 公司;C18固相萃取柱(1000 mg/6 mL)、弱阴离子交换柱Strata®-X-AW(500 mg/6 mL) 美国Phenomenex 公司;Poly-Sery MAX 固相萃取柱(1000 mg/10 mL)、色谱柱(Athena C18-WP, 4.6×250 mm, 5 μm) 上海安谱实验科技股份有限公司;Hypersil GOLD aQ(4.6×250 mm, 5 μm) 美国赛默飞公司;Eclipse XDB-C18(4.6×250 mm,5 μm) 美国安捷伦公司。
1.2 实验方法
1.2.1 标准溶液的配制
1.2.1.1 米酵菌酸标准储备液(1000 μg/mL) 准确称取米酵菌酸标准品10 mg(精确至0.01 mg),用甲醇溶解后,转移至10 mL 容量瓶中,用甲醇定容至刻度。置于2~8 °C 冰箱中避光保存。
1.2.1.2 米酵菌酸标准中间液(10 μg/mL) 准确吸取100 μL 米酵菌酸标准储备液至10 mL 容量瓶中,用甲醇定容至刻度。置于2~8 °C 冰箱中避光保存。
1.2.1.3 米酵菌酸标准工作液 分别移取适量米酵菌酸标准中间液,用甲醇稀释定容,配制成0.01、0.05、0.1、0.3、0.5、1.0、2.0 μg/mL 的标准工作溶液,临用时配制。
1.2.2 样品前处理
1.2.2.1 样品提取 准确称取10 g 样品(银耳样品称取5 g,精确至0.01 g)于50 mL 离心管中,加入10 mL超纯水,20 mL 1%乙酸-乙腈(V/V),4 g 无水硫酸钠、1 g 氯化钠,2 g 无水硫酸镁,迅速振散混匀,涡旋提取30 min,以8000 r/min 离心3 min,取全部乙腈层浓缩至近干,3 mL 甲醇复溶,待净化。
1.2.2.2 样品净化 依次用5 mL 甲醇和5 mL 水活化Poly-Sery MAX 强阴离子交换柱,保持填料湿润。加入100 μL 氨水将3 mL 上样液pH 调节至9~10 过柱,弃去流出液,依次用8 mL 水和8 mL 甲醇淋洗小柱后,弃去淋洗液,最后用8 mL 甲酸-甲醇溶液(4%)洗脱并将洗脱液浓缩近干后用1 mL 甲醇定容,过0.45 μm 有机微孔滤膜,上机测定。
1.2.3 色谱条件 Athena C18-WP 液相色谱柱(4.6×250 mm, 5 μm);流动相为甲醇:1%乙酸-水(80:20,V/V);柱温35 °C;波长269 nm;进样量50 μL。
1.2.4 液相色谱条件的优化
1.2.4.1 检测波长的选择 检测波长对方法的灵敏度、准确度及选择性具有较为关键的影响,本实验对米酵菌酸在190~400 nm 进行全波长扫描,根据“收最大,干扰最小吸”的原则,选择出最佳的检测波长。
1.2.4.2 色谱柱的选择 色谱柱是色谱分离的核心。本实验以甲醇:1%乙酸-水(80:20, V/V)为流动相,在其他参数相同的条件下,分别考察了米酵菌酸标溶液在Athena C18-WP、XSelect HSS T3、Hypersil GOLD aQ 及Eclipse XDB-C18 4 种不同C18 柱上的分离情况,筛选出最佳色谱柱。
1.2.4.3 流动相比例的选择 米酵菌酸具有3 个羧基,在酸性体系可有效抑制米酵菌酸的解离,得到更对称的峰型。因此,本实验在甲醇:1%乙酸-水(80:20, V/V)体系中,采用米酵菌酸标准溶液,分别考察了75%、80%、85% 3 个不同比例甲醇初始流动相的出峰情况,优化出最佳的流动相配比。
1.2.5 样品前处理方法的优化
1.2.5.1 样品提取溶剂的优化 为优化出最佳的样品提取溶剂,本实验采用空白样品加标的方式,选取米粉、银耳、玉米粉、椰子发酵饮料为基质,以加标回收率作为评价指标,分别考察了甲醇、氨水-甲醇-水(1:80:19)、乙腈、1%乙酸-乙腈(1:99, V/V)的提取效果。
1.2.5.2 固相萃取柱的选择 在无净化处理的情况下,杂质干扰大,无法进行准确测定。因此,在其他条件一定的情况下,本实验采用银耳作为研究基质,分别考察了C18、HLB、弱阴离子交换柱strata X-AW,强阴离子交换柱CNW Poly-Sery MAX 4 种固相萃取柱的净化效果,筛选出最佳的净化柱。
1.2.6 米酵菌酸稳定性考察 有关文献提到米酵菌酸在酸性环境及光照情况下不稳定[16],本实验选取米酵菌酸五个浓度(0.1~2.0 μg/mL)的标准品分别置于冰箱避光保存、室内光照3 d、酸性条件保存3 d,在最佳色谱条件下,分别进行上机检测,通过对比其峰面积的变化,考察米酵菌酸稳定性。
1.2.7 方法学考察
1.2.7.1 线性范围及检出限 取系列米酵菌酸标准工作液,经高效液相色谱分析,以米酵菌酸的浓度为横坐标(x),峰面积为纵坐标(y)进行线性回归分析,绘制标准曲线;以3 倍S/N 为检出限,10 倍S/N 为定量限。
1.2.7.2 回收率和精密度 选择米粉、银耳、玉米粉、椰子发酵饮料4 种基质空白样品,加入低、中、高(米粉、玉米粉及椰子发酵饮料:0.030、0.060、0.10 mg/kg;银耳:0.060、0.012、0.10 mg/kg)3 个水平的米酵菌酸标准溶液,每个加标水平进行6 次平行测定,按照1.2.2 节进行前处理后,进行高效液相色谱测定,根据测定结果对其回收率及精密度进行分析。
1.3 膳食风险评估
在食品安全风险评估方面,我国已取得一些成果,但仍处于初级阶段,相关工作仍参考国外膳食暴露风险评估技术[26−27]。本研究参照文献[28]的评价方法,计算米酵菌酸的危害商HQ,当HQ>1 时,表明米酵菌酸的膳食暴露风险较大;反之,当HQ<1 时,则其暴露风险较小。HQ 的具体计算公式如下:

其中“理论含量”采用食品中米酵菌酸的含量,在未检出的样品中,米酵菌酸的含量以上述所建方法检出限的一半计算;参照文献及国家食品安全风险评估委员会的相关规定,米粉、银耳、玉米粉、椰子发酵饮料的每日平均膳食量分别按300、300、300、500 g 计算;平均体重按60 kg 计算;ADI 为每日允许摄入量。
1.4 数据处理
本实验采用SPSS Statistics 22(美国IBM 公司)及Microsoft Excel(美国Microsoft 公司)对检测结果进行相关分析。
2 结果与分析
2.1 液相色谱条件的优化
2.1.1 检测波长的选择 由米酵菌酸的全波长扫描图(图2)可知,在269 nm 处出现最大吸收峰,235 nm次之,根据“收最大,干扰最小吸”的原则,本实验采用269 nm 为米酵菌酸的最佳检测波长。此结果与李红艳等[16]报道的一致。

图2 米酵菌酸吸收光谱图Fig.2 Absorption spectrum of bongkrekic acid
2.1.2 色谱柱的选择 分别采用Athena C18-WP、XSelect HSS T3、 Hypersil GOLD aQ 及 Eclipse XDB-C184 种不同型号的C18柱对米酵菌酸标准溶液进行分析,结果见图3。由图3 可知,米酵菌酸在pH 耐受范围为1.5~10 的Athena C18-WP 液相色谱柱上能够得到最佳的分离效果和稳定的响应值,基质峰不对目标峰造成干扰,因此本实验选择Athena C18-WP 色谱柱进行分离。
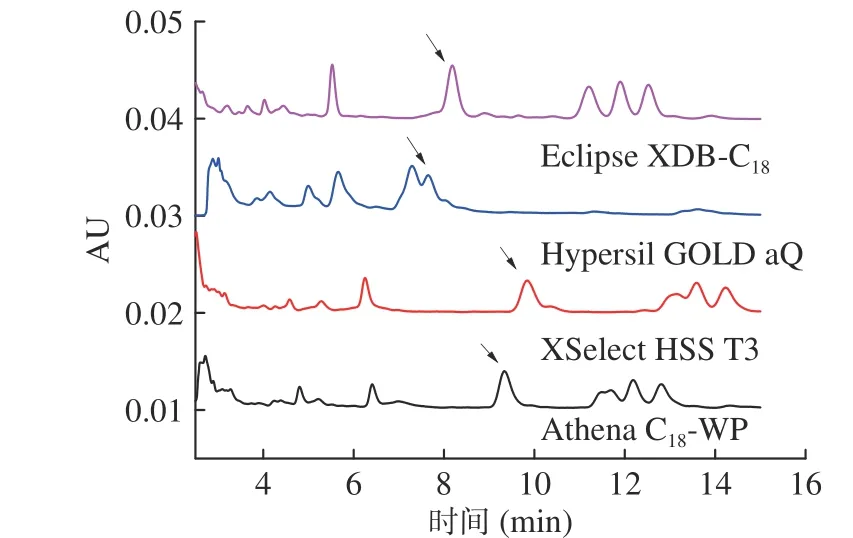
图3 不同C18 柱的分离效果Fig.3 Separation efficiency of different C18 Columns
2.1.3 流动相比例的选择 按照1.2.4.3 节的方法进行试验,结果见图4。由图4 可知,当甲醇比例为75%时,米酵菌酸的出峰时间为13.2 min,且峰宽较大,分离度差;当提高甲醇的比例85%时,米酵菌酸的出峰时间提前至6.2 min,峰形较好,但空白基质样品在6.3 min 附近有干扰峰,不利于准确测定。因此将流动相调节为1%乙酸水-甲醇(20:80)等度洗脱,米酵菌酸在9.5 min 出峰,出峰时间短,且无杂质干扰。
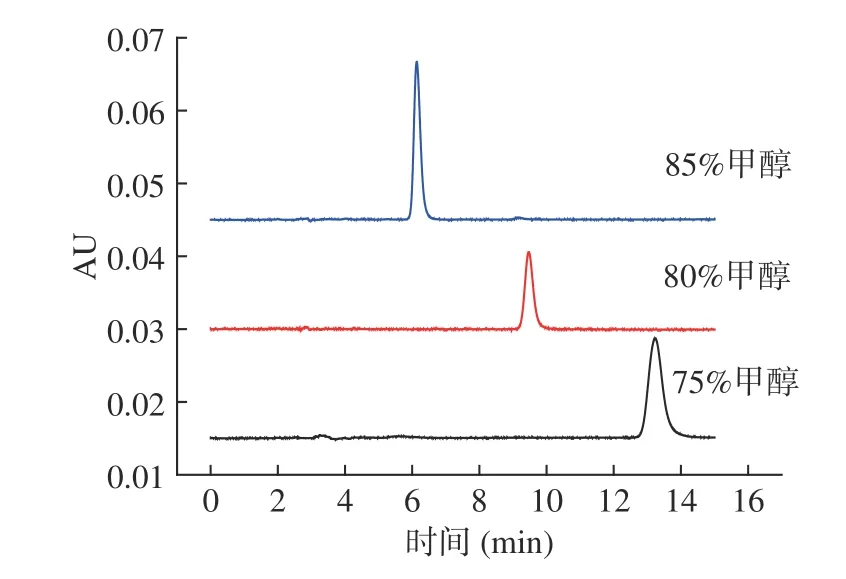
图4 不同流动相中米酵菌酸的标准色谱图Fig.4 Standard chromatograms of BKA in different mobile phase
2.2 样品前处理条件的优化
2.2.1 样品提取溶剂的优化 以米粉、银耳、玉米粉、椰子发酵饮料为基质,分别考察甲醇、氨水-甲醇-水(1:80:19, V/V)、乙腈及1%乙酸-乙腈(1:99,V/V)4 种不同提取溶剂的提取效果,回收率结果见表1。由表1 可知,采用甲醇及乙腈作为提取溶剂时,对4 种基质中米酵菌酸的回收率只有40%~65%,不能满足分析要求;采用氨水-甲醇-水作为提取溶剂时,米粉、玉米粉及椰子发酵饮料中的回收率较高,但是在银耳中的回收率只有60%,而且甲醇和水相不分离导致氮吹浓缩时间较长;另外,对于成分复杂的银耳样品,由于多糖(中性多糖和酸性多糖)含量较高,碱性条件下提取会导致多糖及色素大量溶出,且提取液体较为粘稠,在过柱时容易导致固相萃取柱堵塞或者过载;1%乙酸-乙腈对4 种样品基质中米酵菌酸的提取效果较好,回收率均大于90%,而且,在酸性条件下,米酵菌酸的解离受到抑制,有利于从水相转移到有机相,另外,水相层可以去除部分水溶性干扰物,减少固相萃取柱过载现象,因此,本实验1%乙酸-乙腈(1:99, V/V)作为最佳提取溶剂。
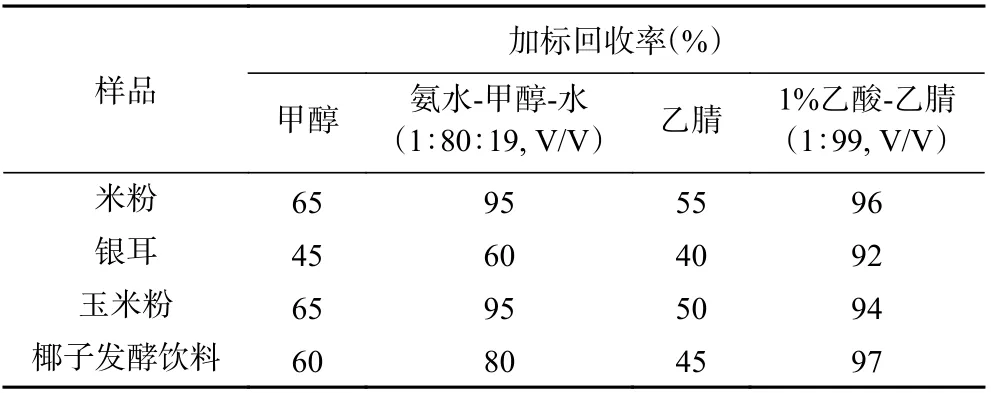
表1 不同提取剂对米酵菌酸的回收率Table 1 The recovery rate of bongkrekic acid by different extractants
2.3 固相萃取柱的选择
本实验考察了C18、HLB、弱阴离子交换柱strata X-AW,强阴离子交换柱CNW Poly-Sery MAX 4 种固相萃取柱的净化效果,结果见图5。由图5 可知,米酵菌酸在C18、HLB 固相萃取柱上几无保留,而在阴离子交换柱strata X-AW,Poly-Sery MAX 有保留,而非文献报道的米酵菌酸于弱阴离子交换柱上无法保留[29]。
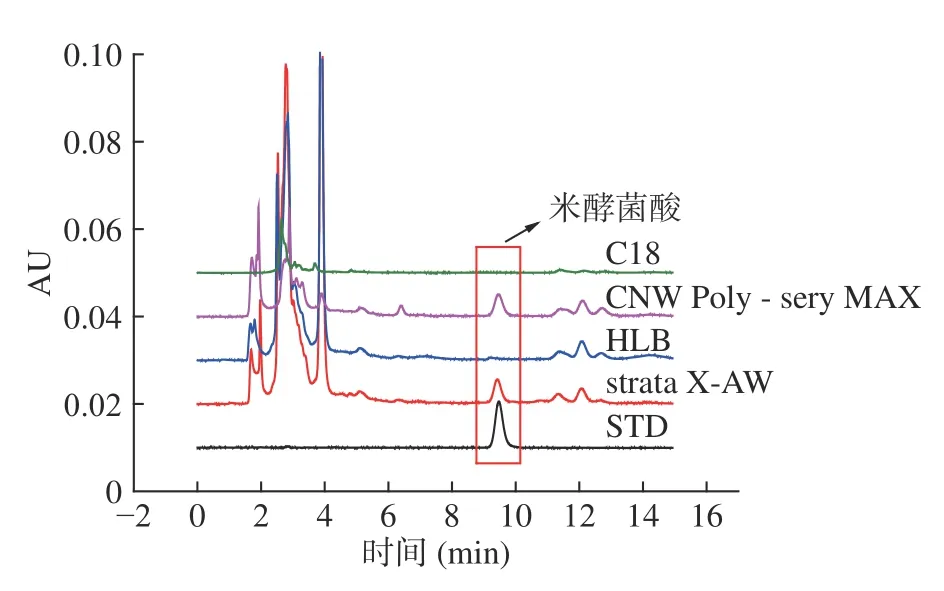
图5 不同型号SPE 小柱净化效果Fig.5 Purification efficiency of different SPE
Strata X-AW 为混合弱阴离子交换柱,含有弱碱性基团,包括伯胺基(-NH2),仲胺基(-NHR),叔胺基(-NR2),在水中能解离出OH-而呈弱碱性,可在中性和酸性条件下使用,其正电基团能与目标化合物的羧基相结合而使得米酵菌酸保留;Poly-Sery MAX 为混合性强阴离子交换柱,含有强碱性季胺基团,pH 耐受范围为2~12,在水溶液中任何pH 条件下都能使硅胶键合相带上正电荷,正电基团能与米酵菌酸的羧基相结合,所以上样液pH 调节至9~10 可使得米酵菌酸完全离子化为阴离子,与强阴离子交换柱的季胺正离子发生高效的离子相互作用而使得米酵菌酸保留。从图5 可知,Strata X-AW 及Poly-Sery MAX柱子的净化效果较好,米酵菌酸出峰位置无其他杂质峰干扰。
另外,本实验对strata X-AW 及Poly-Sery MAX固相萃取柱的加标回收率进行了考察,结果显示,弱阴离子交换柱子strata X-AW 的加标回收率为76%,回收率较低;而Poly-Sery MAX 强阴离子交换柱子的加标回收率达98%,满足检测要求。因此,本实验选用Poly-Sery MAX 固相萃取柱用于样品的净化处理。净化步骤为:依次用5 mL 甲醇和5 mL 水活化小柱;用氨水将待净化液的pH 调节至9~10 过柱,弃去流出液,依次用水和甲醇淋洗小柱,除掉糖类及蛋白质,弃去淋洗液;最后用4%甲酸-甲醇溶液洗脱,并将洗脱液浓缩近干后用1 mL 甲醇定容。
2.4 米酵菌酸稳定性考察
按照1.2.6 节进行米酵菌酸的稳定性试验,结果见图6。由图6 可知,在室内灯光照射与酸性条件下米酵菌酸较为稳定,实验过程使用的酸性条件及室内灯光照射不会影响检测结果。

图6 保存条件对米酵菌酸稳定性的影响Fig.6 Effect of storage conditions on stability of bongkrekic acid
2.5 方法学考察
2.5.1 线性范围及检出限 配制系列标准工作液,在上述最佳分析条件下进行上机检测,以浓度为横坐标,相应的峰面积为纵坐标绘制标准曲线,线性方程为y=1.49×105x−319(R2=0.9999)。结果表明,在0.01~2.0 μg/mL 浓度范围内线性良好。米酵菌酸的检出限(S/N=3)和定量限(S/N=10)分别为2.0 μg/kg和6.7 μg/kg。
2.5.2 回收率和精密度 采用米粉、银耳、玉米粉、椰子发酵饮料4 种不同基质,考察所建方法的回收率及精密度(n=6),结果见表2。由表2 可知,4 种样品平均加标回收率在90.0%~104.0%,相对标准偏差小于5%,结果表明所建方法精密度高,重现性好,可适用于四种高危基质中米酵菌酸的测定。

表2 米酵菌酸回收率及精密度结果(n=6)Table 2 Results of recovery and relative standard deviation(n=6)
2.6 膳食风险评估
为评估广州市在售食品中米酵菌酸膳食风险水平,从广州市市场随机抽取米粉、银耳、玉米粉、椰子发酵饮料样品各10 份,采用所建立的方法进行分析。结果显示,所抽取的40 分样品中,共有8 个样品检出米酵菌酸,含量在10.6~50.8 μg/kg,均低于GB 7096-2014《食品安全国家标准 食用菌及其制品》中0.25 mg/kg 的限量,其中银耳样品检出米酵菌酸最高达50.8 μg/kg,其余样品均未检出米酵菌酸(含量低于2.0 μg/kg)。
目前,国内外对米酵菌酸的ADI 值未有明确的规定。本文根据风险最大化原则,参考GB 7096-2014《食品安全国家标准 食用菌及其制品》规定银耳及其制品米酵菌酸理化指标,中国居民的平均体重以60 kg 计,每日平均膳食量按0.5 kg 计,估算出米酵菌酸的ADI 值为2.08 μg/(kg bw);以随意抽取的40 份样品中米酵菌酸的含量为对象,按1.3 节的方法对米酵菌膳食风险进行评估,结果见表3。由表3可知,米酵菌酸的膳食危害商值为0.0747,远低于临界值1,即米酵菌酸的膳食风险较低,对消费者的膳食安全构成威胁的概率较低。

表3 米酵菌酸膳食风险评估计算结果Table 3 The results of the dietary risk assessment of bongkrekic acid
3 结论
本研究建立了食品中米酵菌酸含量的检测方法。本方法处理简单快速、有机试剂消耗量少、准确性高、适范围较广,方法学指标满足实际检测要求,是现有标准的细化和补充,适用于对米酵菌酸的风险监控,对检测部门开展相应的常规分析检测具有重要的意义;以随意抽取的40 份样品中米酵菌酸的含量为对象,结合膳食风险评估方法,初步评估了米酵菌酸的膳食暴露风险水平。结果显示,米酵菌酸的膳食暴露风险概率为0.0747,远低于临界值1,对一般消费者的健康产生威胁的概率较低。
目前,我国只针对银耳及其制品中米酵菌酸含量作出限量要求,而其他食品中的米酵菌酸含量仍未有明确的规定。因此,建议相关研究人员针对其他食品中米酵菌酸含量进行全面、科学的安全风险评估,并建立相关食品安全标准规范,以加强对米酵菌酸的安全监控,保障消费者的健康。
猜你喜欢
工业水处理(2022年12期)2023-01-05
中国药房(2022年7期)2022-04-14
百姓生活(2021年5期)2021-07-08
化工自动化及仪表(2021年3期)2021-06-04
石油沥青(2021年1期)2021-04-13
食品工程(2020年4期)2021-01-20
中国油脂(2020年3期)2020-04-10
酿酒科技(2019年10期)2019-11-12
海峡姐妹(2018年8期)2018-09-08
家庭医药·快乐养生(2017年6期)2017-06-16
