
甘磷酸胆碱原料药中3种遗传毒性杂质含量测定方法的建立
2021-11-24 19:08张伟奇王倩何燕王雪芹
中国药房 2021年21期
张伟奇 王倩 何燕 王雪芹
中图分类号 R917 文献标志码 A 文章编号 1001-0408(2021)21-2631-04
DOI 10.6039/j.issn.1001-0408.2021.21.12
摘 要 目的:建立氣-质联用(GC-MS)法同时测定甘磷酸胆碱原料药中环氧氯丙烷、缩水甘油、3-氯-1,2-丙二醇等3种遗传毒性杂质的含量。方法:以4批甘磷酸胆碱原料药为检测样品。色谱柱为ZB-WAXplusTM;进样口温度为200 ℃;进样方式为不分流进样;载气为氦气(He),恒流模式;程序升温为初始温度30 ℃保持1 min,然后以30 ℃/min的速率升温至220 ℃并保持5 min;进样量为1 μL。离子源为电子轰击源(EI),电离电压为70 eV,离子源温度为200 ℃;质谱传输接口温度为250 ℃;质谱监测模式为选择离子(SIM);检测特征离子为环氧氯丙烷[质荷比(m/z)49、57、62]、缩水甘油(m/z 31、43、44)、3-氯-1,2-丙二醇(m/z 44、61、79);溶剂延迟时间为3 min。结果:环氧氯丙烷、缩水甘油、3-氯-1,2-丙二醇检测的质量浓度线性范围分别为29.86~746.48、172.91~922.18、21.18~211.85 ng/mL(r均大于0.999 0);检测限分别为19.91、115.27、10.59 ng/mL;定量限分别为29.86、172.91、21.18 ng/mL;精密度(n=6)、重复性(n=6)、稳定性(室温放置12 h,n=8)试验的RSD均小于10%;平均加样回收率分别为93.88%、91.45%、91.86%,RSD分别为5.10%、3.10%、2.49%(n=9)。在4批甘磷酸胆碱原料药中,均未检出上述3种遗传毒性杂质。结论:建立的GC-MS法简单高效、准确度高、重复性好,可用于同时测定甘磷酸胆碱原料药中环氧氯丙烷、缩水甘油、3-氯-1,2-丙二醇等3种遗传毒性杂质的含量。
关键词 气-质联用法;甘磷酸胆碱;环氧氯丙烷;缩水甘油;3-氯-1,2-丙二醇
ABSTRACT OBJECTIVE: To establish GC-MS method for the content determination of three toxic impurities in glycerophosphorylcholine raw materials, such as epichlorohydrin, glycidyl, 3-chloro-1,2-propanediol. METHODS: Four batches of glycerophosphorylcholine raw materials were used as test samples. The determination was performed on ZB-WAXplusTM column, and the injector temperature was 200 ℃; the sample injection adopted splitless injection mode, using helium (He) as carrier gas, in constant current mode; the temperature program for the column was initially heating at 30 ℃ for 1 min, rising to 220 ℃ at a speed of 30 ℃/min then remaining 5 min. The ion source was electrospray ion source (EI), the ion source temperature was 200 ℃, and the ionization energy was 70 eV; transmission interface temperature was 250 ℃, and mass spectrum monitoring mode was selected ion (SIM). Detection ions were epichlorhydrin [mass charge ratio (m/z) 49, 57, 62], glycidyl (m/z 31, 43, 44), 3-chloro-1,2-propanediol (m/z 44, 61, 79). The solvent delay time was 3 min. RESULTS: The linear range of epichlorhydrin, glycidyl and 3-chloro-1,2-propanediol were 29.86-746.48, 172.91-922.18, 21.18-211.85 ng/mL, respectively (all r>0.999 0). The detection limits were 19.91,115.27,10.59 ng/mL, respectively. The limits of quantitation were 29.86, 172.91, 21.18 ng/mL, respectively. RSDs of precision (n=6), reproducibility (n=6) and stability (placed at room temperature for 12 h, n=8) tests were all lower than 10%. The average recoveries were 93.88%, 91.45%, 91.86%, and RSDs were 5.10%, 3.10%, 2.49% (n=9), respectively. In the 4 batches of glycerophosphorylcholine raw materials, three toxic impurities were all not detected. CONCLUSIONS: Established GC-MS method is simple, efficient, accurate and repeatable, and it can be used to determine the contents of three toxic impurities in glycerophosphorylcholine raw materials, such as epichlorohydrin, glycidyl, 3-chloro-1,2-propanediol.
KEYWORDS GC-MS method; Glycerophosphorylcholine; Epichlorhydrin; Glycidyl; 3-chloro-1,2-propanediol
甘磷酸膽碱又称甘磷酰胆碱、甘油卵磷脂,是一种人体内天然存在的水溶性磷脂代谢产物,具有提高记忆力、防衰老、降血脂、保肝、健脑等多种功效[1-2]。3-氯-1,2-丙二醇又称3-氯-1,2-二羟基丙烷,是合成甘磷酸胆碱的起始物料,其可能对人体产生肝肾毒性、免疫毒性、遗传毒性、生殖毒性,并具有致癌性[3-5]。环氧氯丙烷、缩水甘油分别为3-氯-1,2-丙二醇合成的起始物料和合成副产物。其中,环氧氯丙烷又称表氯醇、3-氯-1,2-环氧丙烷,因其具有警示结构——环氧结构,故被视为潜在的遗传毒性杂质,具有潜在的致突变性、致畸性和致癌性[6];缩水甘油又称环氧丙醇,相关动物实验证明其具有致癌作用[7]。目前,对于环氧氯丙烷、缩水甘油和3-氯-1,2-丙二醇常见的检测方法为气相色谱(GC)法和气-质联用(GC-MS)法[7-11]。但目前文献报道的方法多是针对食品和环境中这3种遗传毒性杂质的检测,笔者尚未见甘磷酸胆碱化学原料药中上述3种遗传毒性杂质同时测定的文献报道。由于不同样品的基质和检测要求均有差异,既有文献方法不能满足甘磷酸胆碱原料药中上述3种遗传毒性杂质的检测要求。鉴于此,本研究拟建立GC-MS法同时测定甘磷酸胆碱原料药中环氧氯丙烷、缩水甘油和3-氯-1,2-丙二醇等3种遗传毒性杂质的含量,为该原料药的质量控制提供参考。
1 材料
1.1 主要仪器
Trace DSQ型GC-MS仪(包括FINNIGAN Trace DSQ型质谱仪、Thermo FINNIGAN Trace GC ultra型气相仪、Xcalibur工作站)购自美国Thermo Electron公司;XPE-205型电子天平购自瑞士Mettler-Toledo公司。
1.2 主要药品与试剂
环氧氯丙烷对照品(批号G154861,纯度99.9%)、3-氯-1,2-丙二醇对照品(批号G249657,纯度95.6%)均购自德国Dr.Ehrenstorfer公司;缩水甘油对照品(批号101344-201501,供GC检查用)购自中国食品药品检定研究院;甘磷酸胆碱原料药(批号分别为191001、200101、200102、200103,纯度均为99.8%)购自湖南华纳大药厂股份有限公司;甲醇为色谱纯。
2 方法与结果
2.1 色谱条件
色谱柱为ZB-WAXplusTM(30 m×0.25 mm,0.25 μm);进样口温度为200 ℃ ;进样方式为不分流进样;载气为氦气(He),恒流模式;程序升温为初始温度30 ℃保持1 min,然后以30 ℃/min的速率升温至220 ℃并保持5 min;进样量为1 μL。
2.2 质谱条件
离子源为电子轰击源(EI),电离电压为70 eV,离子源温度为200 ℃;质谱传输接口温度为250 ℃;质谱监测模式为选择离子(SIM);检测特征离子为环氧氯丙烷[质荷比(m/z)49、57、62]、缩水甘油(m/z 31、43、44)、3-氯- 1,2-丙二醇(m/z 44、61、79);溶剂延迟时间为3 min。
2.3 溶液制备
2.3.1 混合对照品溶液的制备 分别取环氧氯丙烷、缩水甘油、3-氯-1,2-丙二醇对照品各约10 mg,精密称定,置于不同10 mL量瓶中,分别用甲醇溶解并定容,摇匀,作为各对照品的单一贮备液①。分别精密量取各对照品的单一贮备液①适量,置于不同量瓶中,用甲醇稀释制成各对照品质量浓度均为10 μg/mL的单一贮备液②。分别精密量取各对照品的单一贮备液②适量,置于同一量瓶中,用甲醇稀释制成环氧氯丙烷质量浓度分别为29.86、199.06、248.83、298.65、398.20、497.65、746.48 ng/mL,缩水甘油质量浓度分别为172.91、230.54、345.82、461.09、576.36、691.63、922.18 ng/mL,3-氯-1,2-丙二醇质量浓度分别为21.18、31.78、42.37、52.96、84.74、105.92、211.85 ng/mL的系列混合对照品溶液。
2.3.2 供试品溶液的制备 精密称定甘磷酸胆碱原料药1 g,置于10 mL量瓶中,用甲醇溶解并定容,摇匀,经0.22 μm滤膜过滤,收集滤液,即得。
2.3.3 空白溶剂 以甲醇为空白溶剂。
2.4 方法学考察
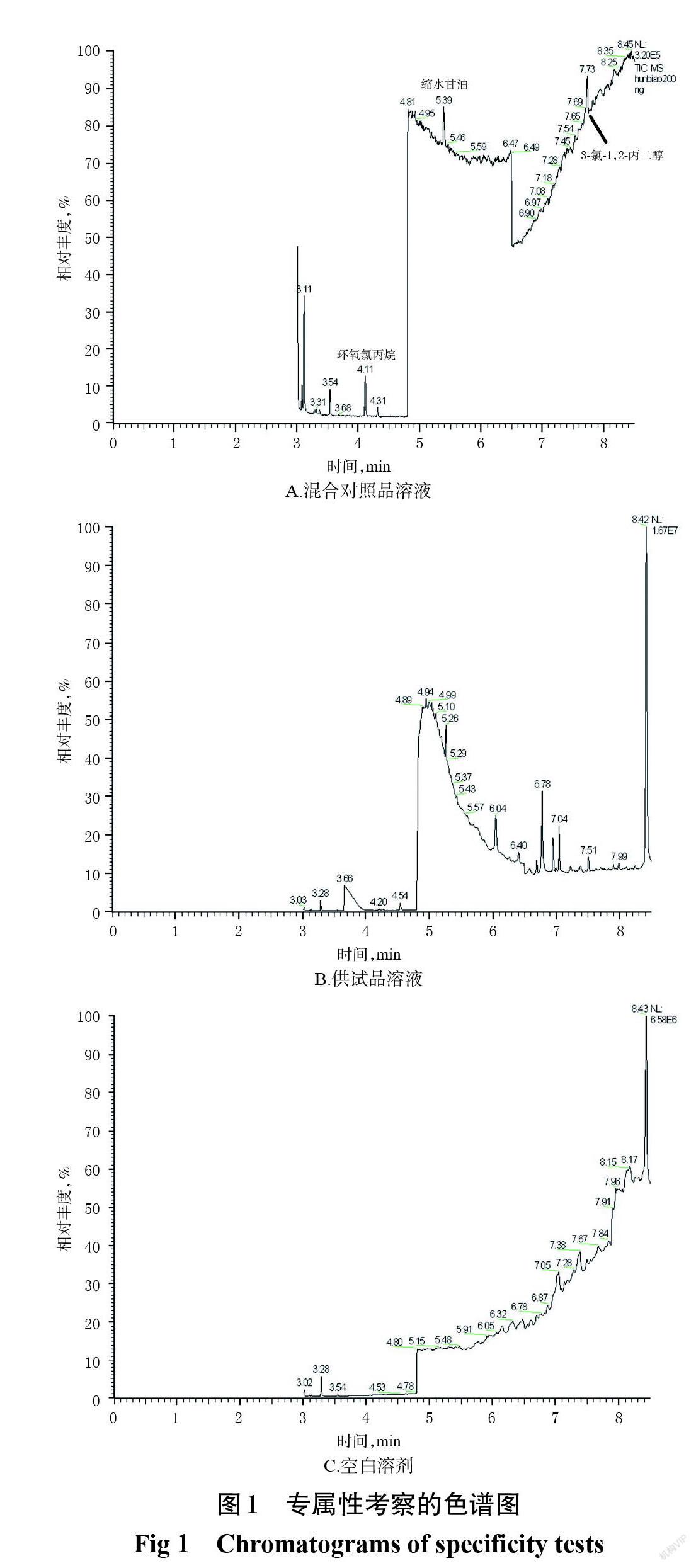
2.4.1 专属性试验 分别精密吸取“2.3”项下环氧氯丙烷质量浓度为248.83 ng/mL、缩水甘油质量浓度为345.82 ng/mL、3-氯-1,2-丙二醇质量浓度为42.37 ng/mL的混合对照品溶液以及供试品溶液(批号200101)和空白溶剂,分别按“2.1” “2.2”项下条件进样测定,记录色谱图。结果显示,环氧氯丙烷、缩水甘油、3-氯-1,2-丙二醇的保留时间分别为4.11、5.30、7.73 min,各待测成分峰均分离良好,无干扰峰,表明该方法的专属性较好。专属性考察的色谱图见图1。
2.4.2 线性关系考察 精密吸取“2.3.1”项下系列混合对照品溶液各1 μL,分别按“2.1” “2.2”项下条件进样测定,记录峰面积。以各待测成分的质量浓度为横坐标(x,ng/mL)、峰面积为纵坐标(y)进行线性回归,得到环氧氯丙烷、缩水甘油、3-氯-1,2-丙二醇的回归方程分别为y=100.19x+265.3(r=0.999 7)、y=94.114x-795.22(r=0.999 5)、y=46.85x-296.5(r=0.999 7)。结果表明,环氧氯丙烷、缩水甘油、3-氯-1,2-丙二醇检测的质量浓度线性范围分别为29.86~746.48、172.91~922.18、21.18~211.85 ng/mL。
2.4.3 检测限和定量限考察 取混合对照品溶液(环氧氯丙烷、缩水甘油、3-氯-1,2-丙二醇的质量浓度分别为9.95、12.81、10.59 μg/mL,制备方法同“2.3.1”项下),用甲醇逐级稀释,然后按“2.1” “2.2”项下条件进样测定,记录峰面积,以信噪比3 ∶ 1、10 ∶ 1分别测得检测限和定量限。结果显示,环氧氯丙烷、缩水甘油、3-氯-1,2-丙二醇的检测限分别为19.91、115.27、10.59 ng/mL,定量限分别为29.86、172.91、21.18 ng/mL。
2.4.4 精密度试验 取“2.3.1”项下混合对照品溶液(环氧氯丙烷、缩水甘油、3-氯-1,2-丙二醇的质量浓度分别为248.83、345.82、42.37 ng/mL),按“2.1” “2.2”项下条件连续进样测定6次,记录峰面积。结果显示,环氧氯丙烷、缩水甘油、3-氯-1,2-丙二醇峰面积的RSD分别为6.1%、3.3%、5.0%(n=6),表明仪器精密度良好。
2.4.5 稳定性试验 取“2.3.1”项下混合对照品溶液(环氧氯丙烷、缩水甘油、3-氯-1,2-丙二醇的质量浓度分别为248.83 、345.82 、42.37 ng/mL),于室温下放置0、1、2、4、6、8、10、12 h时,分别按“2.1” “2.2”項下条件进样测定,记录峰面积。结果显示,环氧氯丙烷、缩水甘油、3-氯-1,2-丙二醇峰面积的RSD分别为8.2%、3.0%、6.5%(n=8),表明该混合对照品溶液在室温下放置12 h内稳定性较好。
2.4.6 重复性试验 取同一批甘磷酸胆碱原料药(批号200101)1 g,精密称定,共6份,分别加入环氧氯丙烷、缩水甘油、3-氯-1,2-丙二醇对照品单一贮备液②各适量,按“2.3.2”项下方法制备含环氧氯丙烷248.83 ng/mL、缩水甘油345.82 ng/mL、3-氯-1,2-丙二醇42.37 ng/mL的供试品溶液,然后按“2.1” “2.2”项下条件进样测定,记录峰面积,采用外标法计算各杂质的含量并计算其回收率。结果显示,环氧氯丙烷、缩水甘油和3-氯-1,2-丙二醇的回收率分别为90.2%、89.9%、90.9%,RSD分别为7.0%、4.1%、7.1%(n=6),表明本方法的重复性较好。
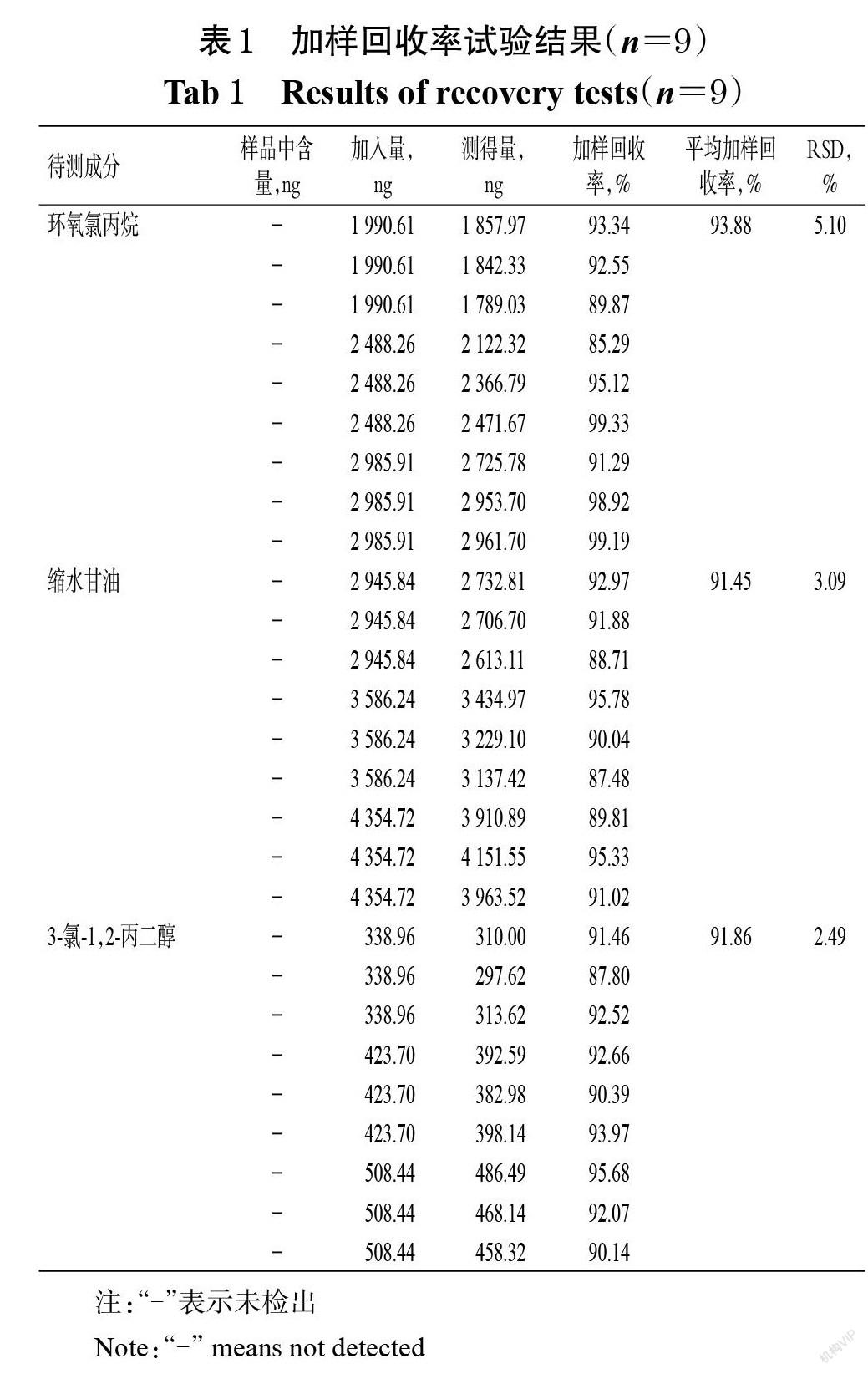
2.4.7 加样回收率试验 取同一批甘磷酸胆碱原料药(批号200101)1 g,精密称定,共9份,置于不同10 mL量瓶中,分别加入各对照品的单一贮备液②适量,并以甲醇为溶剂,分别制备低、中、高浓度的样品溶液(低浓度样品溶液中环氧氯丙烷、缩水甘油、3-氯-1,2-丙二醇的质量浓度分别为199.06、294.58、33.90 ng/mL,中浓度样品溶液中上述3种成分的质量浓度分别为248.83、358.62、42.37 ng/mL,高浓度样品溶液中上述3种成分的质量浓度分别为298.59、435.47、50.84 ng/mL),每个浓度各3份。分别取上述样品溶液,按“2.1” “2.2”项下条件进样测定,记录峰面积,采用外标法计算各杂质的含量并计算其加样回收率。结果,环氧氯丙烷、缩水甘油、3-氯-1,2-丙二醇的平均加样回收率分别为93.88%、91.45%、91.86%,RSD分别为5.10%、3.09%、2.49%(n=9),表明本方法的准确度较好。加样回收率试验结果见表1。
2.5 甘磷酸胆碱原料药中3种遗传毒性杂质含量的测定
取4批甘磷酸胆碱原料药(批号分别为191001、200101、200102、200103),分别按“2.3.2”项下方法制备供试品溶液,然后按“2.1” “2.2”项下条件进样测定,记录峰面积,并采用外标法计算环氧氯丙烷、缩水甘油和3-氯-1,2-丙二醇的含量。结果,4批甘磷酸胆碱原料药中均未检出上述杂质。
3 讨论
3.1 质谱条件的选择
在前期研究中,笔者先在“full scan”扫描模式下对甘磷酸胆碱原料药溶液和环氧氯丙烷、缩水甘油、3-氯-1,2-丙二醇对照品溶液进样后的质谱信息进行采集,并通过与NIST质谱数据库进行比对,确定了环氧氯丙烷、缩水甘油、3-氯-1,2-丙二醇的出峰时间和特征离子(NIST质谱数据库中没有甘磷酸胆碱的相关信息,故未能确定其色谱峰的出峰时间和特征离子):环氧氯丙烷的出峰时间为4.11 min,特征离子m/z为49、57、62;缩水甘油的出峰时间为5.39 min,特征离子m/z为31、43、44;3-氯-1,2-丙二醇的出峰时间为7.73 min,特征离子m/z为44、61、79;溶剂(甲醇)的出峰时间为2.2~2.6 min。然后根据上述结果确定SIM采集模式:从运行3 min起开始检测特征离子m/z为49、57、62的质谱信息,从运行4.8 min起开始检测特征离子m/z为31、43、44的质谱信息,从运行6.5 min起开始检测特征离子m/z为44、61、79的质谱信息,溶剂延迟时间为3 min。接着,笔者以3种杂质质量浓度均为500 ng/mL的混合对照品溶液进样测定,发现各待测成分色谱峰的响应信号和峰形均较好,证实了上述检测方式的合理性。
3.2 色谱条件的选择
在前期研究中,笔者分别考察了不同型号色谱柱(DB-5MS色谱柱和ZB-WAXplusTM色谱柱)的分离效果。结果显示,采用DB-5MS色谱柱和ZB-WAXplusTM色谱柱进行分离时,色谱图中均能显示出3种待测成分的色谱峰信息;但使用ZB-WAXplusTM色谱柱进行分离时,所得色谱图中干扰峰相对较少,而在采用DB-5MS色谱柱进行分离时色谱图中缩水甘油的色谱峰出现了明显拖尾现象,故本研究最终选择ZB-WAXplusTM色谱柱进行后续研究。采用GS-MS法进行含量测定时,为了减少基质及其他组分对质谱检测器的污染,在直接进样方式下的进样量首选为1 μL,当此进样量不能满足检测要求时再增大进样量[12]。笔者在前期预实验中发现,当进样量为1 μL时,各待测成分的响应强度已满足检测要求,故本研究选择进样量为1 μL。
3.3 所建方法的合理性分析
按照人用药品注册技术要求国际协调会议(ICH)和2020年版《中国药典》(四部)关于遗传毒性杂质控制的相关要求[12-14],结合甘磷酸胆碱的临床使用情況(最大日剂量为1.2 g),可计算得环氧氯丙烷的限度为2.46 ppm、缩水甘油的限度为3.56 ppm、3-氯-1,2-丙二醇的限度为0.4 ppm。而本研究所建方法中上述3种杂质的检测限分别为19.91、115.27、10.59 ng/mL(相当于0.019 91、0.115 27、0.010 59 ppm),远低于规定的限度。通过对4批甘磷酸胆碱原料药进行测定,结果均未检出环氧氯丙烷、缩水甘油、3-氯-1,2-丙二醇,表明4批原料中3种遗传毒性杂质的含量低于检测限,满足限度控制要求。
综上所述,本研究建立的GC-MS法简单高效、准确度高、重复性好,可用于同时测定甘磷酸胆碱原料药中环氧氯丙烷、缩水甘油、3-氯-1,2-丙二醇等3种遗传毒性杂质的含量。
参考文献
[ 1 ] 严希康,钱祥云,丁建飞. L-(-甘油磷脂酰胆碱的研发进展[J].上海医药,2015,36(15):68-72,75.
[ 2 ] 卢志花,江磊.甘油磷酰胆碱医药领域专利现状与发展趋势分析[J].中国发明与专利,2018,15(S2):54-58.
[ 3 ] 邢寒竹,方冰,常巧英,等.亚慢性暴露3-氯-1,2-丙二醇对雄性大鼠生殖系统的影响[J].中国食品学报,2019,19(5):39-44.
[ 4 ] 白顺,孙建霞,邹飞雁,等.食品污染物3-氯-1,2-丙二醇毒理作用的研究进展[J].食品工业科技,2013,34(5):358-362.
[ 5 ] 曲花玲,刘晓芳. 3-氯-1,2-丙二醇的毒理学研究进展[J].毒理学杂志,2010,24(2):166-169.
[ 6 ] 赵丽,段毅宏,张瑞雨,等.吹扫捕集-气相色谱串联质谱测定饮用水中的痕量环氧氯丙烷[J].湖北农业科学,2019,58(21):167-169,172.
[ 7 ] 何莲,李怀瑞,朱燮豪,等.测定工作场所空气中缩水甘油的气相色谱法[J].工业卫生与职业病,2017,43(3):225-227.
[ 8 ] 孙筱萍,袁雅文.固相萃取-气相色谱质谱法测定水中环氧氯丙烷[J].河南预防医学杂志,2018,29(9):669-670.
[ 9 ] 戎伟丰,凌伟洁,黄韬,等.气相色谱法同时测定工作场所空气中3种环氧化合物[J].中国职业医学,2014,41(5):573-578.
[10] 薛维丽,由鹏飞,李春焕. GC-MS测定碳酸司维拉姆中环氧氯丙烷的残留量[J].中国现代应用药学,2019,36(9):1089-1091.
[11] 高洁.调味品中3-氯-1,2-丙二醇的GC/MS/MS法检测[J].中国酿造,2013,32(9):145-147.
[12] 国家药典委员会.中华人民共和国药典:四部[S]. 2020年版.北京:中国医药科技出版社,2015:67,527-530.
[13] ICH Expert Working Group. ICH harmonized tripartite guideline:impurities in new drug substances:Q3A(R2)[S]. 2006-06-02.
[14] ICH Expert Working Group. ICH harmonized tripartite guideline:impurities in new drug substances:Q3B(R2)[S]. 2006-10-25.
(收稿日期:2021-05-06 修回日期:2021-09-02)
(编辑:林 静)
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29
中国经济周刊(2021年22期)2021-12-07
科学导报·学术(2020年19期)2020-07-09
环球时报(2020-02-20)2020-02-20
分析化学(2018年12期)2018-01-22
中学科技(2017年11期)2017-12-26
中国经济信息(2017年17期)2017-09-09
中学生数理化·八年级物理人教版(2015年10期)2016-01-04
科学大众(中学)(2009年8期)2009-09-06
祝您健康(1994年11期)1994-12-30
