
铁锰复合金属氧化物的制备及催化燃烧甲苯性能
2021-12-09 00:57江建军喻家欢管国锋
南京工业大学学报(自然科学版) 2021年6期
江建军,喻家欢,邹 建,李 强,管国锋
(1.南通醋酸纤维有限公司,江苏 南通 226008;2.南京工业大学 化工学院,江苏 南京 211800)
化工生产过程中产生的有机废气中含有大量的挥发性有机化合物(VOCs),若不加治理直接排放,会对生态环境和人类健康造成严重影响[1-3]。在所有VOCs处理技术中,催化燃烧法具有处理效率高、无二次污染等优点,已成为研究的热点[4-10]。
催化燃烧的核心在于催化剂的开发。过渡金属氧化物具有多变的价态、低廉的价格、较好的热稳定性等优点,具有替代贵金属催化剂的潜力,已逐渐成为该领域研究的热点[11-15]。常用的非贵金属催化剂有Fe2O3、MnOx、Co3O4和CuO[16-19]等,但是与商用贵金属催化剂相比其活性仍有较大差距。通过构筑复合金属氧化物,可实现催化活性的提升。Wang等[20]采用水热合成法制备了Co3-xMnxO4(x=0.75、1.0、1.5)复合金属氧化物,结果表明通过调控不同n(Co)/n(Mn)比可制得薄片状、花状和蒲公英状的催化剂,Co与Mn之间的相互作用增加了催化剂表面氧物种的数量,提高了催化剂的活性。Chen等[21]将CeO2引入MnOx中,提升催化剂的低温还原性和晶格氧迁移速度,提升了其催化燃烧甲苯的性能,其甲苯转化率为50%时的反应温度(T50)和甲苯转化率为90%时的反应温度(T90)分别为226和239 ℃。而目前通过铁锰之间的相互作用提升催化剂活性的报道较少。
基于上述分析,采用水解-氧化还原法制备了不同铁锰摩尔比的复合金属氧化物催化剂xMn1Fe (x=3、4、5和6),并以甲苯催化燃烧为探针反应,考察了不同铁锰摩尔比、空速对催化剂性能的影响,并通过一系列表征探讨催化活性与物理化学性质之间的关系。
1 实验部分
1.1 主要试剂
九水合硝酸铁、KMnO4、二水合草酸、NaOH和质量分数为50%Mn(NO3)2溶液,上海凌峰化学试剂有限公司,HNO3,南京化学试剂有限公司,甲苯,国药集团化学试剂有限公司,所有试剂均为分析纯。
1.2 催化剂的制备
称取3.792 g (24 mmol) KMnO4和3.232 g (8 mmol) Fe(NO3)3·9H2O溶于200 mL去离子水,剧烈搅拌。4.538 g (36 mmol) H2C2O4·2H2O溶于200 mL去离子水,并逐滴到剧烈搅拌的混合溶液中。6 h之后,将所得深棕色固体过滤,去离子水洗涤,于100 ℃下干燥,400 ℃下焙烧4 h,制得催化剂,记为3Mn1Fe。在此基础上通过加入质量分数为68%HNO3并减少Fe(NO3)3·9H2O用量,调变Mn与Fe的摩尔比,制备出4Mn1Fe、5Mn1Fe和6Mn1Fe。MnO2的制备只加入HNO3,草酸用量不变。
1.3 催化剂的表征
采用日本理学公司的Smartlab TM 9 kW射线仪(CuKα源,辐射波长λ=0.154 18 nm)进行催化剂的XRD分析,扫描范围为10°~80°,扫描速率为10°/min。
采用美国康塔仪器公司的Autosorb EVO进行催化剂N2吸附-脱附表征。实验前,试样在真空中200 ℃预处理4 h,去除表面水分和有机物。
采用HITACHI公司S4800型扫描电子显微镜进行催化剂的形貌分析,工作电压5 kV。
采用美国麦克仪器公司的AutoChem Ⅱ 2920全自动化学吸附仪进行催化剂的H2-TPR测试。试样使用量为30 mg,首先在Ar气氛围200 ℃下预处理2 h,去除催化剂表面吸附的H2O和O2,接着冷却至室温。然后将Ar气变为体积分数为10%H2/Ar混合气,以10 ℃/min的升温速率从室温升到800 ℃。
1.4 催化剂的活性测试
活性测试所用试样质量为100 mg(250~425 μm)与300 mg石英砂(250~425 μm)混合均匀后装填到内径为6 mm的石英管反应器内,两端用石英棉进行固定。反应气体总流量为50 mL/min,对应空速为30 L/(g·h),其中甲苯质量分数为0.1%。测试程序设置升温速率为1 ℃/min,每个取样点稳定15 min并重复取样3次。利用气相色谱测定催化燃烧反应前后甲苯的峰面积,并计算甲苯的转化率。
反应物转化率计算:转化率=(1-反应后色谱积分面积/反应前色谱积分面积)×100%。
应用子系统负责管理持久性数据以及用于显示的临时数据和状态数据,系统内部的数据与系统外部的转换也在这一层次完成,除此之外应用子系统也负责具体应用功能的调用以对来自UI层的功能调用进行响应;同时也控制着所有的全局参数,负责程序的显示配置。该子系统包含命令管理器(Command Manager)、配置模块(Preference)、数据文档(Document)和渲染数据(GL Data)以及接口(Interface)。
2 结果与讨论
2.1 催化剂的表征分析
2.1.1 XRD分析
为了研究试样的晶相组成,进行了XRD表征分析。图1为MnO2、Fe2O3、Cop-3Mn1Fe、3Mn1Fe和xMn1Fe催化剂的XRD图。从图1(a)中可以看出,MnO2在12.8°、18.1°、25.6°、28.7°、37.6°、42.0°、49.8°、56.1°、60.1°、65.7°和69.6°处出现了明显的衍射峰,分别对应于MnO2(JCDPS: 44-0141)的(110)、(200)、(220)、(310)、(211)、(301)、(411)、(600)、(521)、(002)和(541)晶面,这表明通过KMnO4和草酸反应所制得的催化剂为典型的α-MnO2结构。沉淀法制备的氧化铁在24.1°、33.1°、35.6°、40.8°、49.4°、54.1°、62.4°和64.0°处的衍射峰分别对应α-Fe2O3晶相(JCPDS: 33-0664)的(012)、(104)、(110)、(113)、(024)、(116)、(214)和(300)晶面。相较于单一金属氧化物,Cop-3Mn1Fe的XRD图表明Mn物种主要以Mn3O4(JCPDS: 24-0734)形式存在。相比之下,3Mn1Fe的XRD图只出现了1个较宽的峰,说明其结晶度较差。从图1(b)中明显地可以看出,相较于纯的MnO2,5Mn1Fe在2θ=36.7°(对应于(211)晶面)处发生了明显的偏移,其他xMn1Fe也出现了类似现象。这种偏移的原因是Fe3+进入到了MnO2晶格中。同时,从图1(b)中发现,随着Fe含量的增加,MnO2峰强逐渐减弱且峰变宽,主要是Fe的加入降低了MnO2的结晶度,且掺杂量越多,结晶度越低。Fe3+的引入后,MnO2骨架中的Mn4+逐渐转变为Mn3+或Mn2+来维持电荷平衡。通过XRD表征可以看到,通过KMnO4和草酸反应所制得的催化剂为典型的α-MnO2结构,Cop-3Mn1Fe的XRD图则表明Mn物种主要以Mn3O4形式存在。这表明Fe离子的引入使得Mn的平均氧化态下降,氧空位含量增加。平均氧化态的降低意味着Fe的掺杂增强了Mn的Jahn-Teller效应,使得Mn-O键沿C轴拉升变长,Mn-O键减弱,有利于氧空位和缺陷位的生成,暴露更多活性位,增大催化剂的比表面积,从而提升催化剂的催化性能[22-24]。
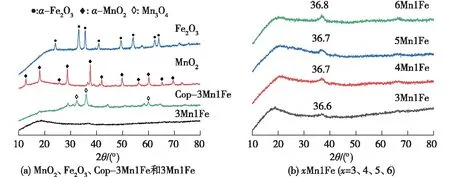
图1 试样的XRD图Fig.1 XRD patterns of samples
2.1.2 N2吸附-脱附分析
为了进一步探索不同方法和不同铁锰摩尔比对催化剂孔结构的影响,对所制备试样进行了N2吸附-脱附表征,结果如图2(a)所示。所有催化剂的N2吸附-脱附等温线均为典型的Ⅳ型等温线,除了MnO2和Cop-3Mn1Fe带有H3型滞后环,其他催化剂都带有H1型滞后环。不同催化剂的孔径分布曲线如图2(b)所示,MnO2和Fe2O3的孔径都集中在12 nm,Cop-3Mn1Fe的孔径分布在4~20 nm,而水解-氧化还原法制备的xMn1Fe复合氧化物的孔径分布在3~7 nm范围内,其中5Mn1Fe的孔径最为集中,说明孔最为规整。
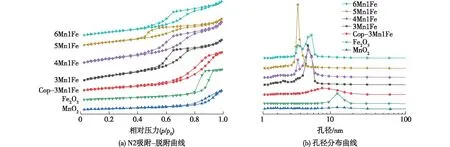
图2 不同催化剂的N2吸附-脱附及孔径分布曲线Fig.2 N2 adsorption-desorption isotherms and pore size distributions of different metal catalysts
不同催化剂的孔容、比表面积和耗氢量如表1所示。MnO2的比表面积为47 m2/g,远小于Cop-3Mn1Fe (114 m2/g)和xMn1Fe (175~219 m2/g),说明引入Fe离子后能进一步增大催化剂的比表面积,且随着x的减小,xMn1Fe的比表面积逐渐增大。较大的比表面积能增强催化剂与反应物之间的接触,提升催化活性。
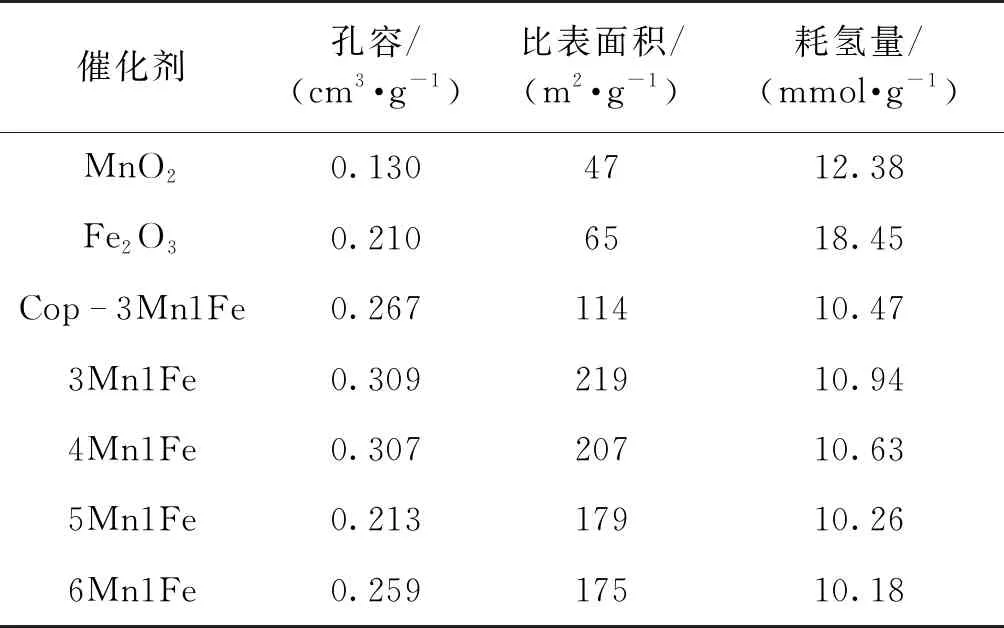
表1 不同催化剂的孔结构性质及耗氢量
2.1.3 SEM分析
为了研究催化剂的形貌,对所制备试样进行了SEM表征,结果如图3所示。图3(a)、3(b)、3(c)、3(d)、3(e)、3(f)、3(g)和3(h)分别表示的是MnO2、Fe2O3、Cop-3Mn1Fe、3Mn1Fe焙烧前、3Mn1Fe、4Mn1Fe、5Mn1Fe和6Mn1Fe不同放大比例的SEM图。从图3(a)中可以看出,KMnO4和草酸反应制得的MnO2呈球形结构,直径约为1 μm,表面粗糙可能是颗粒堆积造成的。图3(b)所示的Fe2O3为颗粒紧密堆积的块状,且形状大小不一。共沉淀法制备的Cop-3Mn1Fe由颗粒无序堆积而成,与纯Fe2O3相似,但表面更为粗糙,故其表面积更大。而水解-氧化还原法制备的不同n(Mn)/n(Fe)比的试样为小球堆积而成。相比Cop-3Mn1Fe要疏松得多,可能是反应过程中产生的CO2起到了扩孔的作用,抑制了颗粒的堆积,增加了催化剂的比表面积。同时,当反应中加入HNO3后,随着n(Mn)/n(Fe)比逐渐增大,催化剂颗粒半径逐渐增大,表面不再光滑而呈现花瓣状。在5Mn1Fe中出现了部分棒状,6Mn1Fe的颗粒状逐渐消失。对比3Mn1Fe焙烧前后的形貌,发现焙烧后3Mn1Fe催化剂形貌无明显变化,说明水解-氧化还原法制备的催化剂结构稳定。

图3 催化剂的不同放大倍率SEM图Fig.3 SEM images of different catalysts at different magnifications
2.1.4 H2-TPR分析
为了研究所制备试样的氧化还原性能,进行了H2-TPR分析。图4(a)为不同方法制得的催化剂的H2-TPR图。MnO2在250~350 ℃范围内不对称还原峰分别对应于MnO2→Mn3O4和Mn3O4→MnO,在较高温度处未出现还原峰。Fe2O3在250~380 ℃和380~650 ℃两个范围内出现还原峰,分别对应于Fe2O3→Fe3O4和Fe3O4→FeO→Fe。Cop-3Mn1Fe和3Mn1Fe在255~386 ℃范围内的还原峰对应MnO2→Mn2O3→Mn3O4和Fe2O3→Fe3O4的连续还原。相比之下,铁锰复合氧化物的体相氧物种还原能力比单一金属氧化物MnO2和Fe2O3更容易,说明共沉淀法和水解-氧化还原法制备的催化剂不是单一氧化物的混合,且3Mn1Fe的低温还原能力更强,可能是均匀分布的Fe和Mn提高了催化剂的氧化还原能力。
xMn1Fe催化剂不同n(Mn)/n(Fe)比对其氧化还原性能的影响如图4(b)所示。所有催化剂出现了相似的还原峰,243~400 ℃范围内的还原峰对应MnO2→Mn2O3→Mn3O4和Fe2O3→Fe3O4的连续还原,500~800 ℃的还原峰对应Fe3O4→FeO→Fe的还原,且5Mn1Fe中Mn的还原温度最低,说明此比例下Mn与Fe的相互作用最强,氧化还原能力较强。如表1所示,xMn1Fe催化剂的总耗氢量随n(Mn)/n(Fe)比的增大由10.94降低至10.18 mmol/g,说明Fe的引入能增加催化剂氧物种的含量。
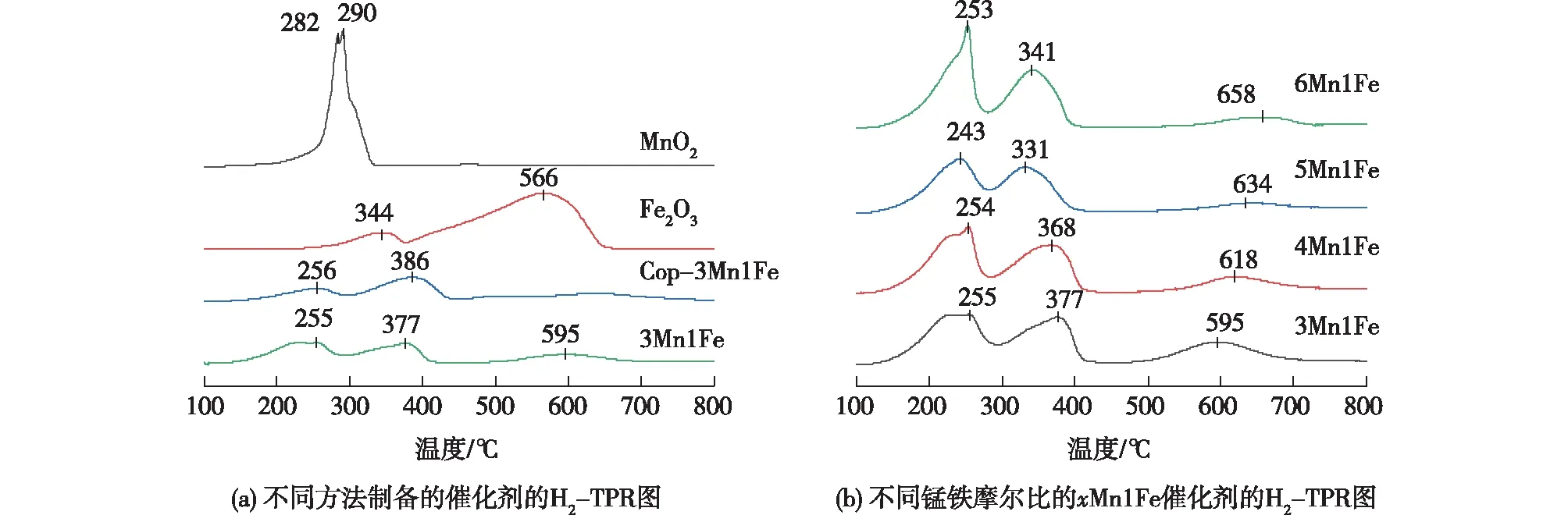
图4 不同方法制备的催化剂和不同锰铁摩尔比xMn1Fe催化剂的H2-TPR图Fig.4 H2-TPR profiles of catalysts prepared by different methods and xMn1Fe with different Mn-Fe molar ratios
2.2 催化剂的性能评价
2.2.1 催化燃烧甲苯性能研究
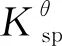
2MnO4-+3H2C2O4+2H+→ 2MnO2+4H2O+6CO2
(1)
Fe3++3H2OFe(OH)3+3H+
(2)
KMnO4与草酸发生反应消耗了Fe3+在水解过程中产生的氢离子,正好促进了Fe和Mn的同时反应,如式(3)所示。
6MnO4-+2Fe3++9H2C2O4→ 6MnO2+2Fe(OH)3+
6H2O+18CO2
(3)
由于不同金属前驱体的沉淀速率和分解速率不同,获得均匀的氧化物是比较困难和不受控制的,这就可能导致金属氧化物以分离相的形式存在,使得催化剂组分布均匀。而水解-氧化还原法则是通过同时进行两个平行反应来合成锰基复合氧化物,元素的分散性较好。Chen等[25-26]的研究工作也证实了这一结论。为了获得活性更优的催化剂,通过调变KMnO4和Fe(NO3)3的量,同时加入适量HNO3得到最佳n(Mn)/n(Fe)比。不同n(Mn)/n(Fe)比的xMn1Fe催化剂活性如图5(b)所示。笔者发现5Mn1Fe的活性最优,T50和T90分别为250和270 ℃,其他比例催化剂活性见表2。对比可得活性由小到大的顺序为3Mn1Fe、4Mn1Fe、6Mn1Fe、5Mn1Fe,与各催化剂的氧化还原能力相一致。通过计算单位催化剂面积、在单位时间内的转化甲苯转化量(比速率),发现比速率的变化趋势与催化剂的性能基本一致,催化剂5Mn1Fe单位面积单位时间内转化甲苯的量最高,达到163 mL/(h·m2)。这表明改性过的催化剂具有较多的活性位点,能促进甲苯的催化转化。此外,表3列出了国内外报道的甲苯燃烧催化剂的性能。相比之下,本文制备的催化剂5Mn1Fe显示出较高的催化活性,具有广阔的应用前景。
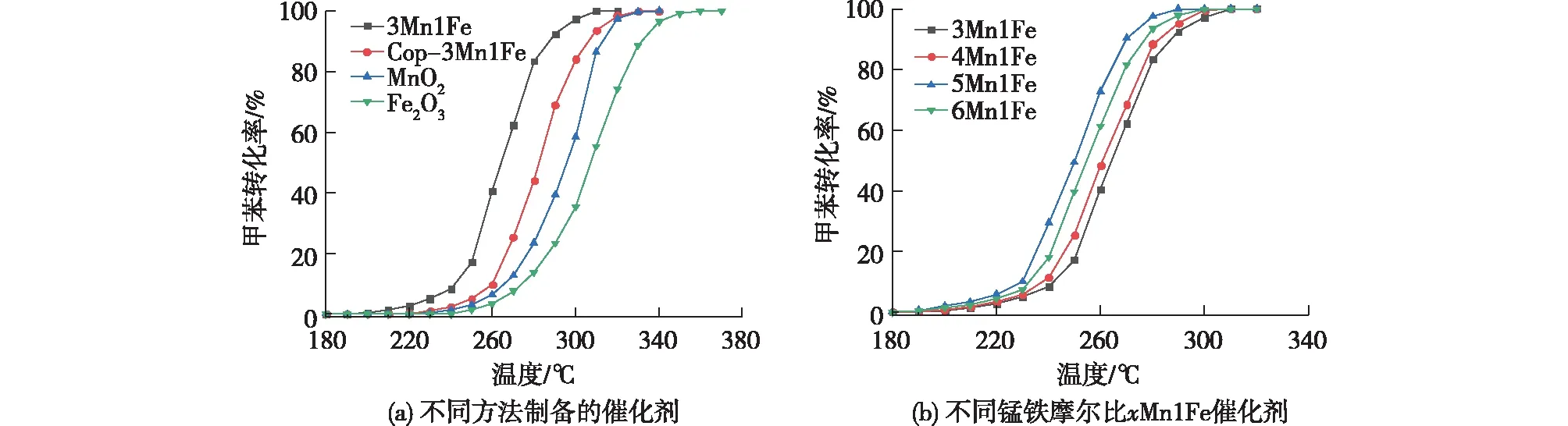
图5 不同方法制备的催化剂和不同锰铁摩尔比xMn1Fe催化剂的活性测试图Fig.5 Activities test diagrams of catalysts prepared by different methods and xMn1Fe binary oxides catalysts with different Mn-Fe molar ratios
2.2.2 空速对5Mn1Fe催化剂性能的影响
实际应用中通常尾气的流量较大,因此需要测试高空速下催化剂的活性。本实验测试过程选取甲苯为VOCs模拟气体,催化剂选用5Mn1Fe催化剂,催化剂用量为100 mg,原料气中甲苯质量分数维持0.1%。

表2 不同催化剂催化燃烧甲苯的活性
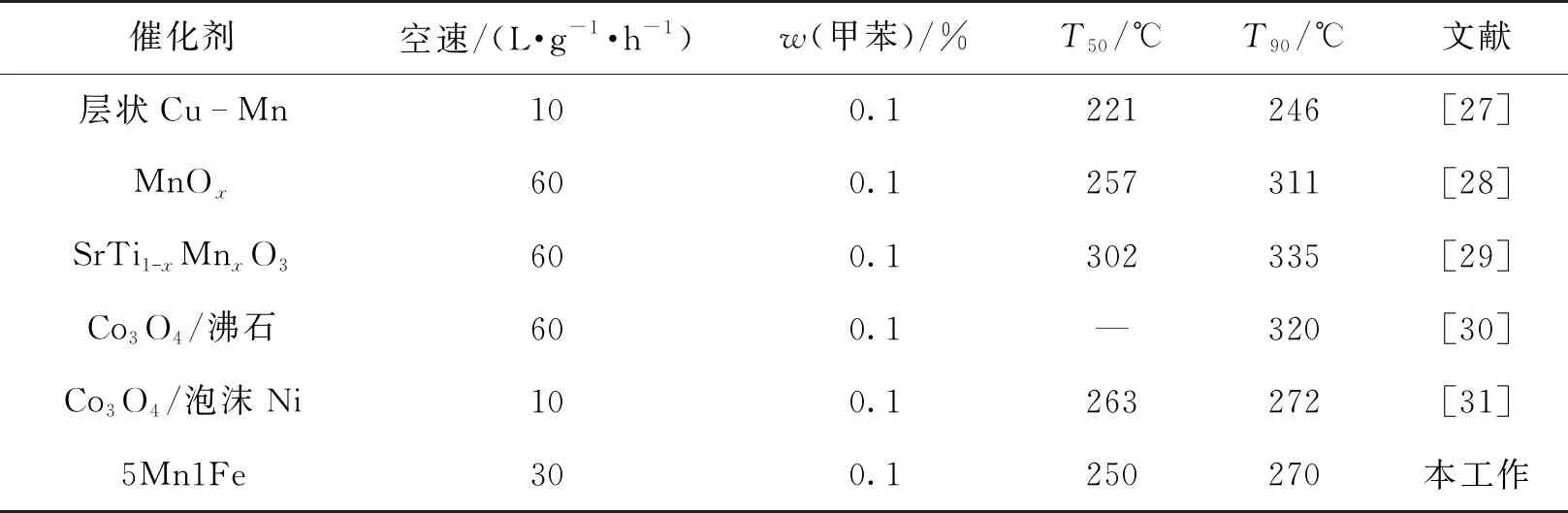
表3 一些甲苯燃烧催化剂的性能比较
5Mn1Fe催化剂在不同空速下催化燃烧甲苯性能结果如图6所示。
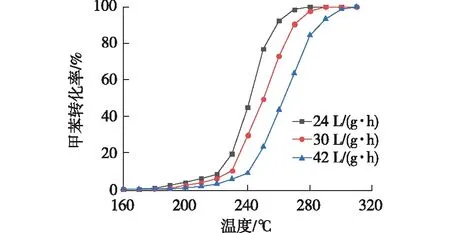
图6 不同空速下5Mn1Fe催化燃烧甲苯转化率曲线Fig.6 Toluene conversion curves by 5Mn1Fe under different WHSVs
从图6中明显可以看出,随着空速从24增大至42 L/(g·h),5Mn1Fe催化活性逐渐降低。相比空速为24 L/(g·h)的情况,空速为42 L/(g·h)条件下的5Mn1Fe甲苯转化对应的T50和T90均升至更高的温度,其中T50升高了22 ℃,T90升高了28 ℃。空速增加导致活性降低的原因可能是高空速下反应气体和催化剂的接触时间缩短,使得反应无法充分进行。
2.2.3 5Mn1Fe催化剂的稳定性研究
催化剂稳定性是评价催化剂能否实际应用的重要指标,因此进行了24 h的5Mn1Fe催化剂稳定性测试。以甲苯为VOCs模拟气体、甲苯质量分数为0.1%、空速为30 L/(g·h)、反应温度为280 ℃,考察甲苯转化率,结果如图7所示。从图7中可以看出,5Mn1Fe催化剂在280 ℃连续反应24 h,甲苯转化率几乎无变化,始终保持大于97%,说明催化剂稳定性较好。

图7 5Mn1Fe催化燃烧甲苯的稳定性测试Fig.7 Long-term stability in the toluene oxidation by 5Mn1Fe catalyst
3 结论
1)由XRD、SEM和N2吸附-脱附可以发现,水解-氧化还原法成功制备了复合铁锰氧化物催化剂,主要为α-MnO2相,呈现出松散堆积的球形颗粒,具有介孔结构,比表面积为179 m2/g。
2)通过H2-TPR分析得出,相较于单一金属氧化物和共沉淀法制备的催化剂,采用水解-氧化还原法制备的铁锰复合金属氧化物的还原温度较低,氧化还原性能较强。
3)水解-氧化还原法制备的催化剂活性明显优于单一金属氧化物和共沉淀法制备的铁锰复合氧化物。当n(Mn)/n(Fe)为5∶1时,催化剂的活性最佳,其T50和T90值最低,分别为250和270 ℃。并且5Mn1Fe催化剂具有很好的稳定性,在280 ℃下连续反应24 h,性能无明显下降。
猜你喜欢
盐科学与化工(2022年6期)2022-06-20
陶瓷学报(2021年4期)2021-10-14
能源工程(2021年2期)2021-07-21
陶瓷学报(2021年1期)2021-04-13
缔客世界(2020年10期)2020-04-10
分析化学(2019年3期)2019-03-30
科学与财富(2017年33期)2017-12-19
中国新技术新产品(2017年3期)2017-03-07
山东工业技术(2016年15期)2016-12-01
中学化学(2015年9期)2016-04-14
