
进行性家族性肝内胆汁淤积症3型合并AIH-PBC重叠综合征1例
2022-01-15 06:54王银玲朱月萍许炜璐黄金龙柴晓哲朱传武
世界华人消化杂志 2022年1期
王 月,王银玲,朱月萍,许炜璐,黄金龙,柴晓哲,李 明,钱 峰,朱传武
王月,王银玲,朱月萍,许炜璐,黄金龙,钱峰,朱传武,苏州市第五人民医院(苏州大学附属传染病医院)感染病科 江苏省苏州市 215131
李明,钱峰,朱传武,苏州市第五人民医院(苏州大学附属传染病医院)肝病科 江苏省苏州市 215131
柴晓哲,苏州市第五人民医院(苏州大学附属传染病医院)消化科 江苏省苏州市 215131
0 引言
自身免疫性肝病包括自身免疫性肝炎(autoimmune hepatitis,AIH)、原发性胆汁性胆管炎(primary biliary cholangitis,PBC)、原发性硬化性胆管炎(primary sclerosing cholangitis,PSC)等.如兼有AIH、PBC或PSC特征者称为重叠综合征,其中以AIH-PBC重叠综合征最为多见.自身免疫性肝病起病隐匿,就诊时往往进展至肝硬化,治疗不及时可短时间内进展至肝功能衰竭.进行性家族性肝内胆汁淤积症3型(progressive familial intrahepatic cholestasis type 3,)是由于基因突变所致的一种遗传性肝病,可引起严重的肝内胆汁淤积,临床预后不良.现将我院收治的1例PFIC3合并AIH-PBC重叠综合征患者的资料报道如下.
1 病例简介
患者,女,32岁.2年前因“肝功能异常伴黄疸11年,加重1月”于2019-07-15首次收住我院感染病科.患者于2008年体检发现肝功能异常,当地医院给与“护肝、降酶”等药物治疗,肝功能异常有所改善.期间2次怀孕,均伴有肝功能异常,当地医院诊断为“妊娠期肝内胆汁淤积症(intrahepatic cholestasis of pregnancy,ICP)”,予对症治疗,分娩后肝功能未恢复正常.2014-03-26复查肝功能:总胆红素(total bilirubin,Tbil) 26 μmol/L、直接胆红素(direct bilirubin,Dbil) 15.8 μmol/L、谷丙转氨酶(alanine aminotransferase,ALT) 255 U/L、谷草转氨酶(aspartate aminotransferase,AST) 212 U/L、碱性磷酸酶(alkaline phosphatase,ALP) 896 U/L、谷氨酰转肽酶(glutamyl transpeptidase,GGT) 605 U/L;B超示肝内钙化灶,脾脏172 mm × 40 mm,间断口服保肝药物治疗.
入院前于2019-06-28在门诊检查肝功能:Tbil 190.4 μmol/L、Dbil 101.1 μmol/L、ALT 118 U/L、AST 114 U/L、ALP 382 U/L、GGT 626 U/L、白蛋白43.0 g/L、球蛋白35.9 g/L;免疫球蛋白G(immunoglobulin G,IgG) 17.4 g/L、免疫球蛋白M(immunoglobulin M,IgM) 3.12 g/L;甲乙丙丁戊型病毒性肝炎血清学标志物均阴性;自身免疫性肝病抗体:抗核抗体(antinuclear antibody,ANA)阳性、抗线粒体抗体(anti-mitochondrial antibody,AMA)等其他自身抗体均为阴性.入院查体:神志清,精神疲软,面色晦暗,肝掌阳性,未见蜘蛛痣,皮肤巩膜中度黄染.腹平软,未见腹壁静脉显露,肝肋下未及,剑突下5 cm,质地中等,无触痛.脾脏肿大,下缘平脐,右侧超过腹中线,质地中等,无触痛.Murphy's征阴性,全腹无压痛及反跳痛,移动性浊音阴性,无肝区叩击痛,双下肢无凹陷性水肿.否认慢性病史及传染病史,无吸烟及饮酒史.已婚,育有2子,子女及父母体健.
入院后化验及检查:肝功能:Tbil 139 μmol/L、Dbil 74.8 μmol/L、ALT 123 U/L、AST 127 U/L、ALP 291 U/L、GGT 512 U/L、白蛋白37.9 g/L、球蛋白32.9 g/L;血常规:白细胞3.0×10/L、红细胞3.23×10/L、血红蛋白103 g/L、血小板85×10/L;IgG 13.4 g/L、IgM 2.65 g/L;EB病毒、巨细胞病毒抗体、弓形虫抗体、HIV抗体、梅毒抗体均阴性;凝血功能、甲状腺功能、肿瘤指标均未见异常;α1-酸性糖蛋白、铜蓝蛋白均正常;自身免疫性肝病抗体:ANA阳性、抗核点型抗体阴性、抗核糖核蛋白抗体阴性、AMA-M2阴性、抗SP-100抗体和抗GP210抗体均阴性,IgG4阴性;无创肝纤维化检测:LSM 12.6 kPa,CAP 198 db/mL.腹部B超:肝损害声像图,肝内增强回声(结石?钙化灶?),门静脉内径10 mm,脾静脉内径8 mm;胆囊轻中度炎性改变;脾肿大.
入院后初步诊断考虑为AIH-PBC重叠综合征可能,给予熊去氧胆酸、甘草酸制剂等药物治疗,同时进一步给予检查.2019-07-22肝穿刺活检,病理结果提示:轻中度界面炎,汇管区局部桥接坏死,汇管区及纤维隔内有较多淋巴细胞、少量浆细胞浸润,可见小胆管损伤,部分汇管区与小动脉伴行的小胆管减少或消失;肝细胞轻度水肿,少许气球样变及脂肪变性,可见点状坏死,玫瑰花结样肝细胞,汇管区周围肝细胞内见淤胆性色素颗粒.结论:符合原发性胆汁性胆管炎叠加自身免疫性肝炎(PBC+AIH),改良Scheuer评分:G3S3(见图1).
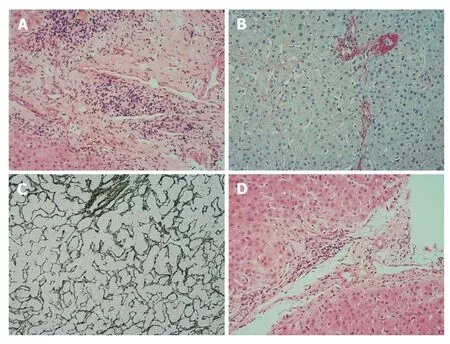
图1 肝活检病理结果.A:汇管区大量炎性细胞浸润;B:天狼星红染色示桥接纤维化;C:网染见肝板网状支架局部紊乱;D:汇管区未见与小动脉伴行的小胆管.A,C,D:×100;B:×40.
2019-07-28腹部磁共振成像(magnetic resonance imaging,MRI)和磁共振胰胆管造影(magnetic resonance cholangiopancreatography,MRCP)提示:(1)脾脏肿大;(2)肝脏形态异常,考虑肝硬化可能;(3)右肝管及部分分支显影不清,肝左叶远端部分肝内胆管略显扩张;(4)肝脏多发小囊肿;(5)胆汁粘稠.
2 诊断与治疗
综合病史及入院后检查以及肝穿刺病理结果,AIH综合诊断积分系统评分为19分,简化诊断标准评分为7分,AIH可以确诊;ALP和GGT显著增高,IgM水平增高,肝组织学显示小胆管损伤、部分汇管区小胆管减少或消失,PBC诊断明确.因此,患者诊断为AIH-PBC重叠综合征.遂予“泼尼松20 mg qd,熊去氧胆酸(ursodeoxycholic acid,UDCA)250 mg tid”联合治疗.治疗期间复查血生化指标,ALT、AST、ALP及GGT等有所下降,但均未恢复正常;Tbil无明显下降,维持在(100-160) μmol/L之间;IgG恢复正常,IgM 2.65 g/L,仍高于正常值上限.
由于患者脾脏肿大明显,显著异于常见的AIH-PBC重叠综合征患者,且患者也无疟疾、伤寒、血液系统疾病及心血管疾病史,因此,于2019-08-05建议患者做黄疸相关的基因检测.基因测序高通量检测结果提示:基因存在大片段杂合缺失突变,见图2.

图2 高通量基因测序提示ABCB4基因存在大片段杂合缺失突变.
患者给予“甲泼尼龙片联合UDCA”治疗,于2019-08-10带药出院.结合患者病史、临床症状和体征、相关实验室及影像学检查、黄疸相关基因测序结果及诊疗经过,出院诊断为:(1)PFIC3合并AIH-PBC重叠综合征;(2)肝硬化(代偿期).出院时肝脏Child-Pugh评分6分,对疾病发展的不良预后也对患者和家属做了交代.考虑此疾病为常染色体隐性遗传性疾病,建议其直系亲属完善相关基因检测,同时建议患者早做肝移植准备,患者因经济原因暂缓.
3 结果和随访
2020-04-21患者因“上消化道出血”第二次收住我院肝病科.呕血2次,鲜红色,量约300 mL.期间检查肝功能:Tbil 63 μmol/L、Dbil 53.9 μmol/L、ALT 36 U/L、AST 79 U/L、ALP 267 U/L、GGT 446 U/L、白蛋白30.0 g/L、球蛋白34.8 g/L;凝血功能:凝血酶原时间(prothrombin time,PT)14.8秒,凝血酶原活动度(prothrombin activity,PTA)64.8%;血常规:白细胞6.83×10/L、红细胞2.18×10/L、血红蛋白51 g/L、血小板117×10/L;IgG 15.6 g/L、IgM 2.08 g/L;甲胎蛋白(alpha-fetoprotein,AFP)正常;自身免疫性肝病抗体:ANA阳性、AMA-M2阳性、抗SP-100抗体和抗GP210抗体均阴性.经过止血、输血、抑酸护胃、补充白蛋白等治疗,出血控制,病情好转.因处于新冠肺炎疫情期间,胃镜检查未开放,故未行胃镜检查,于2020-05-01带药出院,嘱咐定期随访.出院诊断为:(1)PFIC3合并AIH-PBC重叠综合征;(2)肝硬化(失代偿期);(3)上消化道出血.
2021-06-04患者因再次呕血第三次住院,消化科给与内镜下精细精准食管胃底静脉曲张断流术(endoscopic selective variceal devascularization,ESVD)止血治疗,镜下见食管静脉重度曲张并破裂出血.入院后检查肝功能:Tbil 218.0 μmol/L、Dbil 176.8 μmol/L、ALT 29 U/L、AST 58 U/L、ALP 121 U/L、GGT 70 U/L、白蛋白26.3 g/L、球蛋白31.8 g/L;凝血功能:凝血酶原时间19.1秒,凝血酶原活动度 42.9%;血常规:白细胞6.91×10/L、红细胞2.93×10/L、血红蛋白94 g/L、血小板136×10/L;IgG 18.7 g/L、IgM 2.88 g/L;血氨38 μmol/L;AFP正常,CA125 232.2 U/mL;腹水常规提示为漏出液,培养无细菌生长.腹部B超示肝硬化、脾肿大、门静脉内径10 mm,脾静脉内径9 mm,腹腔积液.经止血、补充白蛋白、利尿等治疗,出血停止、腹水消退,于2021-07-09出院.2021-08-11随访,肝功能:Tbil 487.4 μmol/L、Dbil 367.9 μmol/L、ALT 131 U/L、AST 171 U/L、ALP 97 U/L、GGT 63 U/L、白蛋白31.8 g/L、球蛋白33.6 g/L;凝血功能:凝血酶原时间 15.3秒,凝血酶原活动度60.1%;血常规:白细胞11.82×10/L、红细胞3.38×10/L、血红蛋白113 g/L、血小板73×10/L.患者病情的逐步进展也使其认识到肝移植为最终治疗方案,目前在当地医院住院,等待行肝移植治疗.
4 讨论
AIH表现为血清IgG水平升高,ANA或抗平滑肌抗体(antismooth muscle antibody,ASMA)高滴度阳性,组织学特征为肝实质细胞炎症,胆管损害较轻.PBC是一种自身免疫介导的、针对胆管上皮细胞的自身免疫性肝病,其病理损害特征是小叶间胆管非化脓性破坏性胆管炎,亚临床期可长达10-15年,但在出现临床症状后可迅速进展至胆汁淤积、肝硬化和肝功能衰竭.AIH-PBC重叠综合征占所有PBC患者的5%-15%,兼有AIH和PBC的血清生化学特征,主要包括血清ALT、AST、ALP、GGT升高,同时伴有IgG、IgM水平不同程度的升高;自身抗体中多出现ANA、ASMA、AMA或抗SP-100、抗GP210抗体阳性.
本例患者有反复肝功能异常史10余年,曾在多家医院就诊,筛查临床常见的病毒性肝炎血清学指标、自身免疫性肝病相关抗体等,均未见明显异常,故一直未能明确诊断.在本院首次住院期间,检查肝功能酶学指标、IgG和IgM水平、肝病相关自身抗体,并结合肝穿刺病理学检查,患者的诊断符合AIH-PBC重叠综合征的诊断标准.关于AMA-M2,患者首次住院前和住院期间检查均为阴性,但在第二次住院期间复查AMA-M2为阳性,说明在高度怀疑自身免疫性肝病时,相关自身抗体的检测需要多次复查,有时最好应用不同的检测试剂复查.在确诊后,给与患者糖皮质激素联合熊去氧胆酸治疗,肝功能指标有所改善,但总体效果欠佳.特别是患者脾脏显著肿大,显著异于常见的AIH-PBC重叠综合征患者.因此,为了解是否存在其他黄疸相关的肝病,进一步完善了血清IgG4检测以及遗传代谢性肝病的基因测序.结果发现基因存在大片段杂合缺失突变,提示患者同时合并遗传性胆汁淤积性肝病.
PFIC是一组常染色体隐性遗传性疾病,其特征是由于胆汁合成和运输障碍引起的肝内胆汁淤积,多发生在婴幼儿时期,少部分类型可于成年后出现肝硬化.根据已知的致病基因,PFIC分为3型.PFIC3的特征是基因发生突变,导致其编码的多药耐药蛋白3(multidrug resistance protein 3,MDR3)缺失,使胆汁磷脂酰胆碱分泌所需要的脂肪酶缺乏或功能异常.MDR3蛋白是肝细胞毛细胆管面上的磷脂转运酶,负责将磷脂从肝细胞内转运至毛细胆管面,在胆管中与胆盐混合形成微粒排泌至胆汁中.基因突变导致MDR3蛋白表达下降或缺失,引起胆汁中磷脂比例降低或缺乏,不能形成磷脂与胆盐的混合微粒,造成胆盐游离,对胆管细胞和肝细胞发生毒性去垢作用.持续的细胞损伤及炎症因子浸润最终导致胆汁淤积、小胆管增生、广泛门管区纤维化和肝硬化,最后发展为终末期肝病.PFIC1型和2型分别由ATP8B1和ABCB11基因突变引起,PFIC3型由基因突变引起.PFIC3型区别于1型和2型的临床特征是血清GGT水平升高,本病例符合PFIC3型的诊断标准,但是因为合并有AIH-PBC重叠综合征,使得病情更为复杂,也更为少见.
本病例是由于血清胆红素、ALT、AST、ALP和GGT水平长期异常,影像学显示肝内外胆管通畅,经过自身免疫性肝病相关抗体检测,以及肝组织活检病理学检查,首先确诊了AIH-PBC重叠综合征.但是在糖皮质激素联合UDCA治疗下,肝功能指标改善并不显著,并且患者的脾脏显著肿大也不能完全以AIH-PBC重叠综合征的诊断得到合理解释,因此,考虑是否合并有引起肝内胆汁淤积的其他肝病存在,比如PFIC或者良性复发性肝内胆汁淤积症(benign recurrent intrahepatic cholestasis,BRIC)等.通过基因测序检查,确认患者同时合并PFIC3.目前报道的基因突变有很多种,包括错义、无义及缺失突变等,基因突变的类型与病情严重程度有关,严重者在儿童早期即可发展为肝功能衰竭.另外,基因突变也可引起诸如ICP、胆石症、PBC、药物性肝内胆汁淤积症等疾病.本例患者在首次入院的诊治过程中,各项检查支持AIH、PBC和PFIC3各自的诊断标准,因此,PFIC3合并AIH-PBC重叠综合征是明确的.从随后的病情演变看,肝病进展较快,肝移植治疗是最后的治疗手段.
5 结论
目前,综合利用生化学、血清学、影像学、组织学、基因学等方法,能够让绝大多数病因不明的肝病患者获得确诊.但是,类似于本例因为肝功能反复异常,辗转就医,且疗效不佳,病情进展而久未获得确诊的疑难复杂性肝病在临床上也并不鲜见.AIH-PBC重叠综合征诊断相对容易,糖皮质激素等免疫抑制剂联合UDCA治疗对大多数患者是有效的,但对于少数治疗效果不佳的成人胆汁淤积性肝病,要考虑到合并PFIC3等遗传性胆汁淤积性肝病的可能.如果不仔细分析病情,很容易导致漏诊.本病例在明确AIH-PBC重叠综合征的诊断后,再确诊了PFIC3,虽不能为患者提供有效的治疗方案,但有助于了解疾病发展的不良预后,减少患者因为诊断不明而反复就医的成本,同时也为临床医生诊治疑难性肝病提供了参考.
猜你喜欢
基层中医药(2022年7期)2022-11-17
中国现代医生(2022年19期)2022-11-04
传染病信息(2022年2期)2022-07-15
保健医苑(2022年5期)2022-06-10
中国典型病例大全(2022年12期)2022-05-13
现代临床医学(2022年1期)2022-02-12
智慧医学(2021年1期)2021-09-10
珠江水运(2021年15期)2021-08-29
水能经济(2017年6期)2017-10-19
中国民族民间医药·下半月(2014年2期)2014-09-26
