
2-甲基丁酰-辅酶A脱氢酶缺乏症患者ACADSB基因变异分析
2022-01-19 00:41李莉朱丹丹张凤
海南医学 2022年1期
李莉,朱丹丹,张凤
1.泸州市妇幼保健院儿科,四川 泸州 646000;2.绵阳市第三人民医院(四川省精神卫生中心),四川 绵阳 621000
2-甲基丁酰-辅酶A脱氢酶缺乏症(2-methylbutyryl-CoA dehydrogenase deficiency,2-MBAD)是 由ACADSB基因变异引起的异亮氨酸分解代谢受损的隐性遗传代谢性疾病,也称短/支链酰基辅酶A脱氢酶缺乏症(short/branched chain acyl-CoA dehydrogenase deficiency,SBCADD)[1]。在新生儿疾病筛查开展前,多由出生后出现喂养困难、低血糖或在儿童期出现发育迟缓、智力障碍、癫痫、肌肉萎缩、肌张力减退、自闭症等临床表现进行检查而确诊,但随着串联质谱的开展,大部分患者在出现临床表现前已被确诊[2]。本研究对3例串联质谱筛查时发现异戊酰基肉碱(C5)升高患者进行基因检测,确诊为2-MBAD,并进行ACADSB基因变异分析,以明确其致病原因,为家系的遗传咨询提供依据。
1 资料与方法
1.1 一般资料 选取2018—2020年泸州市妇幼保健院新生儿疾病筛查串联质谱发现异戊酰基肉碱(C5)升高疑似2-MBAD的3例患者,收集串联结果及临床资料,并对其进行遗传代谢性疾病相关基因检测。
1.2 方法
1.2.1 DNA提取 经监护人签署知情同意书、并经医院伦理委员会批准后,抽取患者及其父母静脉血2 mL,EDTA抗凝,送北京博奥医学检验所,使用天根生化科技(北京)有限公司的微量样品基因组DNA提取试剂盒提取基因组DNA。
1.2.2 高通量测序及Sanger测序验证 采用探针法靶向捕获3 877个已知的疾病基因,配合Illumina Next 500测序平台获得测序数据,筛选出变异位点。再用Sanger测序方法验证患者父母。
1.2.3 变异位点的生物信息学分析 用SIFT、PolyPhen-2、MutationTaster等软件对候选点变异进行位点保守性预测、蛋白功能预测;用GnomAD、ExAC和1000Genomes数据库对变异位点进行人群频率注释;用ClinVar数据库对变异位点的致病性判读结果进行注释。选择GnomAD等数据库中东亚人群频率低于0.01,并且位点保守性强、对蛋白结构或功能影响剧烈,ClinVar中为非良性变异,且基因对应的临床表型与患者表型吻合的基因变异位点查阅文献进行变异位点的致病性分析。
2 结果
2.1 临床资料
2.1.1 病例1 男,58 d,因新生儿疾病筛查C5升高就诊,患者系G2P1,足月顺产,出生体质量3.3 kg,无窒息、抢救病史,患者生后精神奶量可,大小便正常。父母体健,非近亲结婚,否认家族遗传病史。查体:体质量4.1 kg,身长53 cm,头围39 cm。前囟1.8 cm×1.4 cm,平软,神清,皮肤弹性可,心肺腹无异常;四肢肌力、肌张力正常,神经系统检查无异常。辅助检查:血常规、肝肾功能、心肌酶谱、电解质、血糖、血氨、血乳酸未见异常。串联结果:C5 0.56μmol/L,C5/C2 0.04,C5/C3 0.40(第一次);C5 0.60μmol/L,C5/C2 0.04,C5/C3 0.30(第二次)(参考范围C5 0~0.4μmol/L,C5/C2 0~0.04,C5/C3 0~0.4)。
2.1.2 病例2 女,21 d,因新生儿疾病筛查C5升高就诊,患者系G1P1,足月顺产,出生体质量2.8 kg,无窒息、抢救病史,患者生后精神奶量可,大小便正常。父母体健,非近亲结婚,否认家族遗传病史。查体:体质量3.2 kg,身长51 cm,头围36 cm。前囟1.8 cm×1.5 cm,平软,神清,皮肤弹性可;心肺腹无异常;四肢肌力、肌张力正常,神经系统检查无异常。辅助检查:血常规、肝肾功能、心肌酶谱、电解质、血糖、血氨、血乳酸未见异常。串联结果:C5 7.94μmol/L,C5/C2 0.19,C5/C3 1.82(第一次);C5 8.81μmol/L,C5/C2 0.91,C5/C3 5.99(第二次)。
2.1.3 病例3 女,44 d,因新生儿疾病筛查C5升高就诊,患者系G1P1,足月顺产,出生体质量3.1 kg,无窒息、抢救病史,患者生后精神奶量可,大小便正常。父母体健,非近亲结婚,否认家族遗传病史。查体:体质量4.0 kg,身长51.6 cm,头围38 cm。前囟1.8 cm×1.4 cm,平软,神清,皮肤弹性可;心肺腹无异常;四肢肌力、肌张力正常,神经系统检查无异常。辅助检查:血常规、肝肾功能、心肌酶谱、电解质、血糖、血氨、血乳酸未见异常。串联结果:C5 1.37μmol/L,C5/C2 0.14,C5/C3 0.93(第一次);C5 1.54μmol/L,C5/C2 0.04,C5/C3 0.35(第二次)。
2.2 基因检测结果 病例1,ACADSB基因外显子5,c.655G>A(p.V219M)(纯合变异),分别来自父母;病例2,ACADSB基因外显子10,c.1165 A>G(p.M389V)(纯合变异),分别来自父母;病例3,ACADSB基因外显子5,c.655G>A(p.V219M)(杂合变异),来自母亲,外显子10,c.1165 A>G(p.M389V)(杂合变异),来自父亲,如图1。通过Phyre2功能软件可知655位缬氨酸及1165位蛋氨酸位于相对保守区域,如图2。c.655G>A(p.V219M)及c.1165A>G(p.M389V)位点突变没有导致氨基酸之间的氢键改变,如图3。

图1 ACADSB基因变异测序图(箭头所指为变异位点)
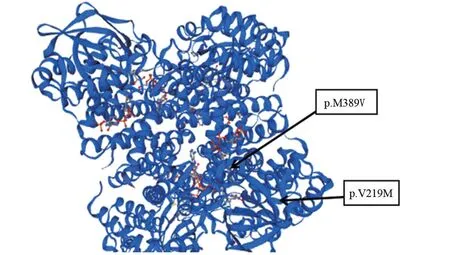
图2 ACADSB基因变异图
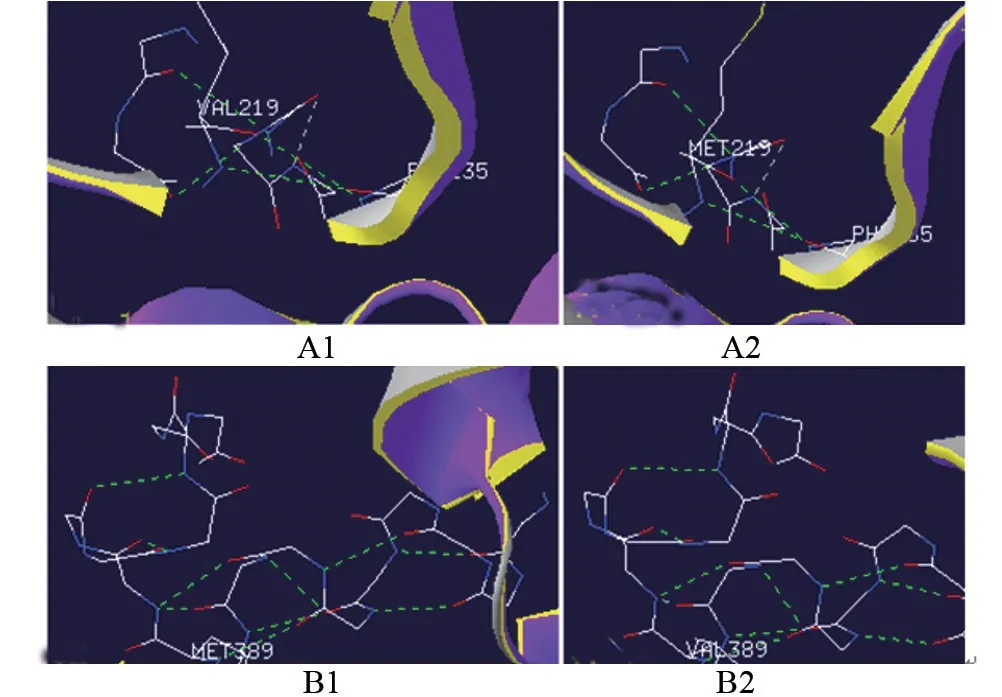
图3 变异位点氢键连接
3 讨论
异亮氨酸作为支链氨基酸(branched-chain amino acids,BCAAs)之一,在生长、免疫、蛋白质代谢、脂肪酸代谢、葡萄糖转运等全身生理功能中起着至关重要的作用[3]。研究发现,异亮氨酸可以诱导宿主防御肽(β-防御素)的表达,从而调节宿主的先天免疫和适应性免疫,进而改善免疫系统,包括免疫器官、细胞和活性物质。此外,异亮氨酸可通过增加抗菌素的表达,恢复某些病原体对人和动物健康的影响[4]。异亮氨酸代谢异常可致多种代谢性疾病[5],2-MBAD就是其中之一,如图4所示。2-MBAD是由ACADSB基因变异所致,发病率极低,目前全球发病率不详,但在东南亚苗族和苗族后裔中较为常见,该区域发病率高达1/250~1/500,有文献报道,我国泉州市发病率为1/30 379[1]。ACADSB基因(NG_008003.1)位于10号染色体(10q26.13),共11个外显子,编码432个氨基酸的2-甲基丁酰辅酶A脱氢酶,该酶在异亮氨酸(Isoleucine)代谢途径中发挥重要作用,该基因缺陷导致2-甲基丁酰肉碱、2-甲基丁酰甘氨酸等代谢物异常蓄积[6]。
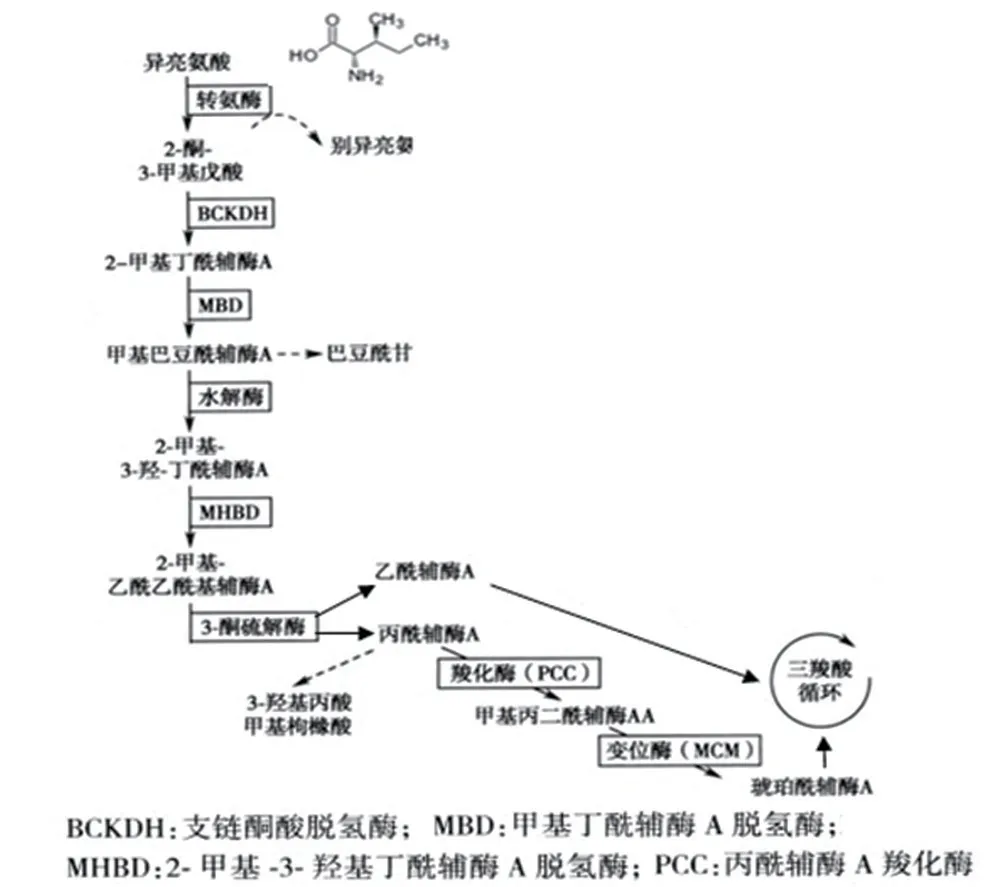
图4 异亮氨酸代谢
研究发现,苗族华人大部分2-MBAD病例无临床症状,可能与其他短支链酰基辅酶A活性重叠,避免异亮氨酸有害代谢物的积累[7],但在感染和应激情况下可能发生新生儿危象,严重者发生发育迟缓、智力障碍、癫痫、肌肉萎缩、肌张力减退、自闭症等[8]。
ACADSB基因c.655G>A(p.V219M)的变异不属于多态性变化,发生频率极低,目前尚无报道。该位点变异219位缬氨酸与235位苯丙氨酸之间的氢键没有发生改变,但与220位的蛋氨酸形成二肽改变。有研究发现,缬氨酸-蛋氨酸经历电子吸收,产生带有SO键的阳离子,该阳离子可以在缬氨酸-蛋氨酸中性子上去质子化,产生的中性自由基。但蛋氨酸-蛋氨酸二肽不能产生中性自由基,因此导致2-甲基丁酰辅酶A脱氢酶活性减低[9]。
c.1165A>G(p.M389V)为热点变异位点,位于亲水口袋中[10],在美国威斯康星州该位点纯合变异占所有变异位点中的1.3%,杂合子变异占21.8%[11];而在中国泉州该位点变异率占所有变异位点中的33.3%[1]。该位点由蛋氨酸变异为缬氨酸,氨基酸之间的氢键没有改变,但分子遗传学分析发现该位点使ACADSB基因在转录过程中外显子10跳跃,导致2-甲基丁酰辅酶A脱氢酶活性减低[7]。
2-MBAD的治疗资料少且无统一的治疗共识,目前治疗措施以改善临床症状、低蛋白饮食及补充肉碱为主,避免禁食。
综上所述,本文3例患者在新生儿疾病筛查时发现串联质谱异常,基因检测诊断2-MBAD,予以低蛋白饮食及补充肉碱,目前尚未发生发育迟缓、智力障碍、癫痫、肌肉萎缩、肌张力减退等,但后期是否发生,需长时间追踪随访。c.655G>A(p.V219M)目前暂无报道,新变异的检出丰富了基因变异谱,为家系的遗传咨询提供了依据。
猜你喜欢
生物信息学(2022年3期)2022-11-12
中老年保健(2022年4期)2022-08-22
中国饲料(2022年5期)2022-04-26
全科护理(2022年3期)2022-02-18
西南农业学报(2021年10期)2021-12-14
中国康复(2021年6期)2021-11-30
猪业科学(2020年12期)2021-01-09
华声文萃(2019年4期)2019-09-10
文萃报·周二版(2019年10期)2019-09-10
家庭百事通·健康一点通(2017年8期)2017-08-18
